
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2020.07
Abstract:
All eukaryotic cells, including the pancreatic acinar cell, tightly control organization of their cytoplasm which in large part relies on the cytoskeleton. The cytoskeleton is comprised of three filament types: microtubules (tubulin), intermediate filaments (keratins), and microfilaments (actin). In addition to characteristics common to all cells, the pancreatic acinar cell is a polarized epithelial cell with a vectorial arrangement of the secretory machinery and apically-directed protein secretion, with important roles of the cytoskeleton in this polarity. The most investigated of the cytoskeleton elements in the acinar cell are actin-based microfilaments which are involved in regulated protein secretion. The development of the fluorescent Lifeact probe for realtime visualization of actin dynamics in live cells makes this an exciting time for the field. The dynamic rearrangement of the actin cytoskeleton associated with protein secretion is the focus of this review.
I. Introduction
The actin cytoskeleton that is ubiquitous in nonmuscle cells is largely made up of b-actin and g-actin, the products of the ACTB and ACTG1 genes, respectively (2). These two actins differ by only 4 amino acids but they appear to have different functions, as mutations in the two genes have very different effects (2). In the mouse acinar cell, Actb mRNA is expressed more than 2-fold greater than Actg1 (Table 1); presumably their protein levels match this expression although this has not been measured in the pancreas. In gastric parietal cells, b-actin is more localized to the apical cortical cytoskeleton whereas g-actin is more localized along the basolateral plasma membrane (BLPM) (48). If the same is true in the acinar cell, then b-actin is more likely to be important in protein secretion which is also localized to the apical surface of the cell. In the acinar cell, g-actin may be involved in the BLPM blebs that form under supraphysiologic stimulation. From here on, actin will be referred to as if it were a single gene/protein.
Results from studies of the actin cytoskeleton in the exocrine pancreas are the primary source for this review. However, there are many gaps in our knowledge regarding exocrine pancreas function. When data from pancreas are lacking, results from other cell types that can reasonably be assumed to be true of the acinar cell are briefly discussed. It should be noted that even though there are commonalities shared by all cells with a regulated secretory pathway [e.g., regulation by small GTPases and use of SNAREs to mediate granule/vesicle - plasma membrane (PM) recognition and fusion (19)], there can be important differences.
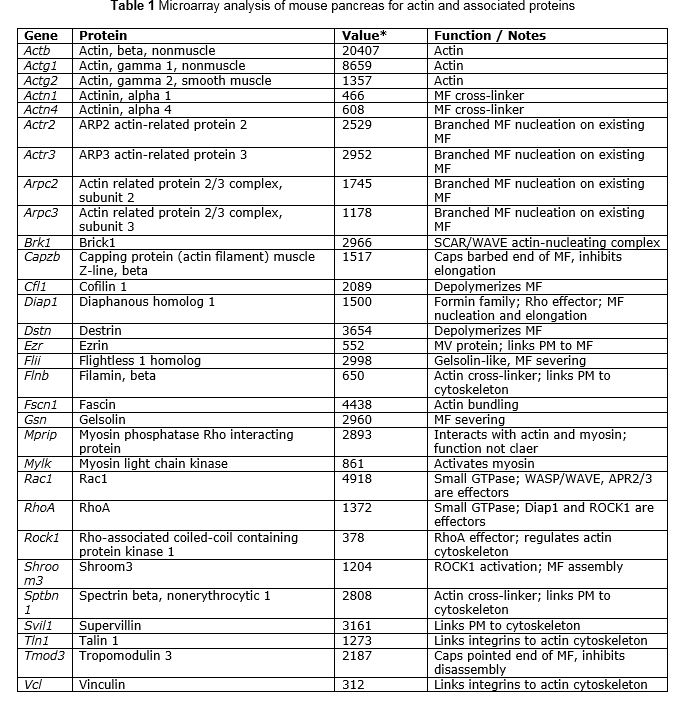
From microarray analysis of mouse pancreas (http://www.ncbi.nlm.nih.gov/geo/, accession GDS567). Data are means from 3 wild type young adult mice. See (18) for details. * Value: "normalized signal count data, reflecting the relative measure of abundance of each transcript" as defined on the GEO website. This list is not comprehensive. Many actin cytoskeletal genes were expressed below detection limits, or were not represented on the microarrays.
For example, the role of actin coating of granules/vesicles during the content release process may serve different functions related to the speed of product release required (see 4 E. Dynamics of acinar actin in the stimulated state below).
II. Actin Basics
Actin is a 42 kDa protein that exists in two distinct forms in the cell: monomeric (globular or G-actin) and polymeric (filamentous or F-actin). Actin monomers have the inherent behavior of self-assembly into filaments and prevention of inappropriate polymerization is controlled by several G-actin binding proteins that stoichiometrically outnumber G-actin by more than 3-fold 47). However, in general the majority of a cell's actin is in the form of F-actin, estimated to comprise ~75% of the total (47). The structure of F-actin microfilaments (MF) is a polarized double helix such that the barbed (+) end is where new monomers are primarily added and the pointed (-) end is where F-actin primarily releases actin monomers. Actin is an ATPase, and for G-actin the ATP-bound form predominates and is thus primed for addition to an actin filament. Once added to the filament, the ATPase is activated and ATP is hydrolyzed to ADP.
F-actin is highly dynamic and there are numerous proteins that regulate the assembly and disassembly of MF. Interrogation of a published microarray study done in the author's lab(18)for actin cytoskeleton elements was performed and is presented in Table 1. This yielded a number of genes, the most highly expressed of which not surprisingly were b- and g-actins, as well as several proteins known to bind to or regulate dynamics of actin. Many of these proteins directly interact with actin and are thus called actin binding proteins (ABP) (42). The majority of genes on this list and their functions were discovered using other cell systems and have not been investigated in the pancreatic acinar cell. Microfilament (MF) structures in cells may appear to be highly stable structures, but they are constantly undergoing growth at the (+) end and disassembly at the (-) end, a process called treadmilling (38). Thus, the apparent stability of MFs is actually due to a dynamic equilibrium. It has been estimated that the ~1 mm long MFs of the intestinal microvillus turns over every 20 min by treadmilling, and a similar rate is also believed to be true of the terminal web (TW) that underlies the apical plasma membrane (APM) (9). In addition to proteins that induce disassembly of MFs, there are numerous actin-binding proteins that regulate nucleation of new fibers and growth of existing fibers. Actin can exist as higher order bundles of multiple MFs in linear arrays as well as branched MF structures, with accessory proteins providing lateral cross-links (42).
III. Actin Localization and Dynamics in the Acinar Cell
A. Methods to Visualize F-actin
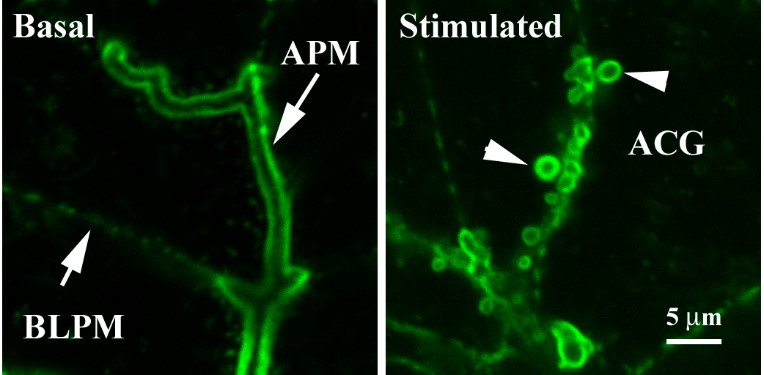
Most studies of actin in the acinar cell have been of MFs and usually after chemical fixation of the cells in either the unstimulated basal state, with a secretory stimulus, and/or after a specific experimental manipulation of the cells. The most commonly used technique is fluorescently tagged phalloidin, a fungal toxin with a high affinity for F-actin (see Pancreapedia articles and images therein: Rab3; Visualization of exocytosis in pancreatic acinar cells by fluorescence microscopy; Rab27). An example of FITC-phalloidin labeling of mouse acini from the author's lab is shown in Figure 1, which shows the MFs of acinar cells under basal and stimulated conditions. Phalloidin is a stabilizer of F-actin MFs so when used in living cells it perturbs the actin cytoskeleton. This property limits use of phalloidin to static imaging of F-actin after fixation, and precludes its use to visualize dynamics of actin in live cells where other techniques are used (see below).
Figure 1. FITC-phalloidin staining of F-actin in mouse acinar cells under basal and stimulated conditions. Mouse acini were incubated without (Basal) or with 1 M carbachol (Stimulated). The cells were fixed in suspension with 2% formaldehyde, 0.1% saponin to permeabilize cell membranes, and 1 mM MgCl2 and 5 mM EGTA to stabilize F-actin. The cells were then stained with 0.6 M FITC-phalloidin and imaged on a confocal microscope, collecting 0.5 μm optical sections. These images are single optical slices. The acinar lumen is bounded by the APM (left panel), under which is the strongly labeled TW. The BLPM (left panel) is much weaker stained than the APM. After stimulus, there are actin coated granules (ACG) along the APM (right panel, arrowheads) and the TW is more weakly stained. [These images were preliminary data for the work presented in (37).]
F-actin can also be recognized in standard transmission electron microscopy as electron dense ~6 nm diameter filaments (smaller than microtubules, ~24 nm; and intermediate filaments, ~10 nm).
Until recently, MFs have been difficult to visualize in living cells without perturbing the structure and function of the cytoskeleton. Adding tags such as green fluorescent protein (GFP) to b-actin perturbs the actin cytoskeleton into which it is incorporated [discussed in (14)]. A newer approach is to make chimeric fluorescent proteins from ABPs. Using such an approach the Lifeact reporter was made, which consists of a 17 amino acid actin binding domain from the ABP140 protein, fused to GFP (31). This chimeric protein has a 30-fold greater affinity for F-actin than G-actin, was found to not interfere with known activities of the actin cytoskeleton, and it faithfully reproduces known F-actin staining patterns in a variety of cell types even under dynamic situations where the actin cytoskeleton is being remodeled (14). This construct was later used to make a transgenic mouse expressing the fluorescent F-actin reporter in all tissues, called the Lifeact mouse (32).
G-actin can also be visualized using fluorescently tagged DNase I, which binds to both G- and F-actin, with higher affinity to the former (21), although an analysis of DNAse I inhibition by highly purified G- and F-actin concluded that both forms have similar affinity for DNase I (25). An example of fluorescent DNase I staining of G-actin in the acinar cell shows a diffuse cytoplasmic distribution which is not very informative (27). Visualization of G-actin has not been used to study the actin cytoskeleton of the acinar cell to any extent. Nor has there been systematic investigation of G-actin ABPs in the acinar cell.
B. Acinar Cell Actin in the Resting State
The single largest pool of F-actin in the acinar cell is in the TW (4), also called subapical actin or cortical actin (Figure.1, left panel).

Empty fields indicate data was not reported for that parameter. Abbreviations: ACG: actin-coated granules; BDM: 2,3-butadione monoxime; CA: constitutively active; Cyto: cytochalasin; DN: dominant negative; Lat: latrunculin; TW: terminal web
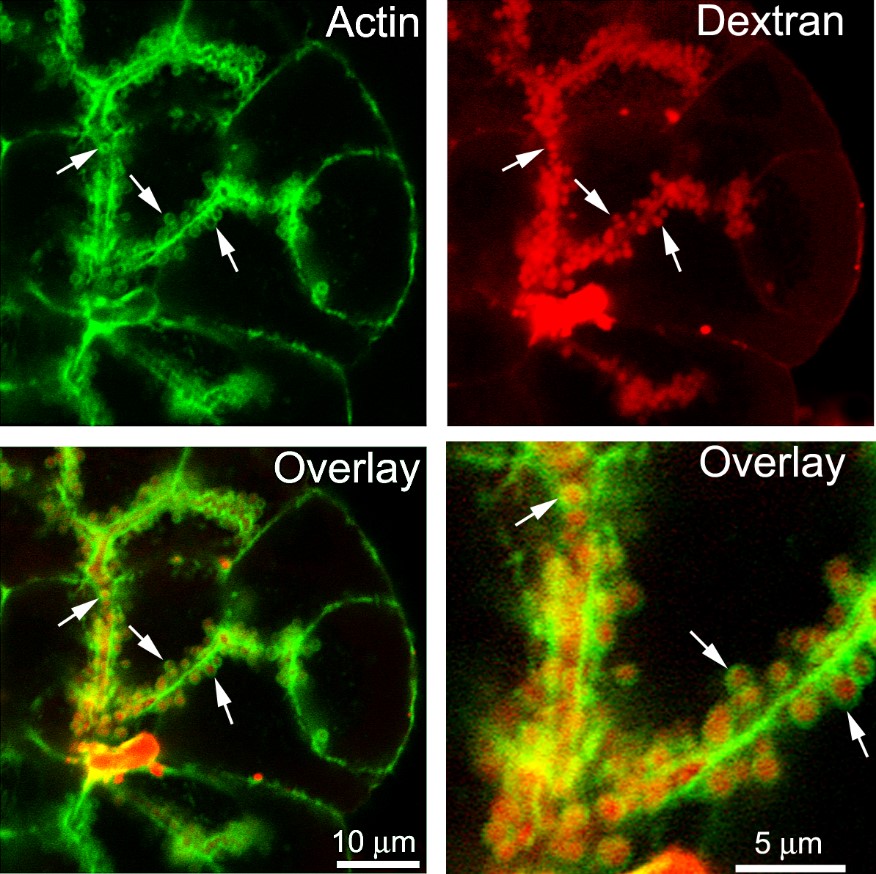
Figure 2. ACG are open to the lumen and accumulate fluorescent dextran from the medium. Acini were stimulated in the presence of fluorescent lysine-fixable dextran (red) and then fixed and costained with FITC-phalloidin, and imaged on a confocal microscope. The enlarged image (lower right) clearly shows that ACG also have fluorescent dextran in their lumina, demonstrating access of the ZG lumen to the medium during secretory stimulation. [Courtesy of John A. Williams, Unpublished]
The TW is a dense arrangement of MF that forms a continuous sheet-like network ~200 nm thick (13) under the APM. Among ABPs, members of the red blood cell spectrin family cross-link TW MFs and also link the TW to perpendicular MF that extend into the microvilli (MV), as shown in intestinal epithelium (16). As compared to the villus epithelium of the small intestine where MV are best characterized, the acinar cell MV are relatively sparse. A second actin-based structure is a ring of MFs that are attached to the adherens junctions near the apical surface and encircle each acinar cell. Because this is a ring-shaped structure it is less easily visualized than the sheet-like TW. There are also recently-described MF bundles that project perpendicularly from the TW into the cytoplasm that are called actin cables (15). Lesser amounts of MFs are found along the BLPM of the acinar cell (Figure 2, left panel). Because of the large amount of F-actin in the TW, it can be difficult to visualize the other sites of MFs when using fluorescent labeling approaches. For example, the recently described actin cables that run perpendicular to the TW in acinar cells are not observed using phalloidin staining, but are visualized using a chimeric ABP domain fused to GFP (Lifeact, see below) (15).
C. Effects on Secretion of Pharmacological Agents and Molecular Approaches that Perturb the Actin Cytoskeleton
There is a large data set about the acinar cell actin cytoskeleton obtained using a variety of approaches. Because the actin cytoskeleton is so important to eukaryotic cells, there are numerous naturally occurring substances produced by a variety of organisms that interfere with the organization of actin, and many of these are available as research tools. Also, investigators have produced constitutively active (CA) and dominant negative (DN) constructs to stimulate or inhibit upstream pathways that control the actin cytoskeleton. A summary of these toxins and reagents and their effects on the actin cytoskeleton and acinar cell amylase release is given in Table 2. Some of the reported effects of a particular agent are contradictory but these are likely attributable to differences in concentration of the agent (e.g. latrunculins have been used over the wide range of 0.3 mM to 100 mM), time of exposure, and temperature at which the experiment was performed [discussed in (7)]. The importance of a specific agent's concentration is illustrated by the finding discussed below that a small perturbation of the actin cytoskeleton increases amylase release, whereas stronger perturbation blocks amylase release (26). The data covered here attempts to give the consensus of the available data.
D. Signals that Affect the Actin Cytoskeleton
The principal mechanism of acinar cell stimulation begins with ligand binding to membrane receptors coupled to G proteins that feed into several pathways. With respect to the acinar cell actin cytoskeleton, the known pathways are activation of the small GTPases RhoA (via Ga13) and Rac1 (via Ga13 and Gaq) (35). These activated small GTPases in turn activate their downstream effectors such as Diap1 and ROCK1; and WASP/WAVE and ARP2/3, respectively, that lead to actin cytoskeleton remodeling.
As discussed below, a major downstream effector of Rho activation is the formin family member Dia. In drosophila, the formin Diaphanous is activated by the small GTPase Rho, resulting in nucleation and formation of linear F-actin filaments that comprise the TW of epithelial cells (23). Although not yet experimentally verified, Rho activation in the mammalian pancreas is expected to activate mDia1 and it has been shown in mouse acinar cells that constitutively active (CA)-mDia1 leads to F-actin cables perpendicular to the apical PM (15). It was suggested these F-actin cables serve to recruit zymogen granules (ZG) through the TW to the apical membrane during secretory stimulation (15). Consistent with the requirement for apical MF in secretion, protein release into the tracheal lumen of Drosophila lacking Dia, which also lack TW F-actin, was absent. A notable difference between Drosophila and mammalian acinar cells is that Dia in drosophila is required for formation of the TW of epithelia (23); whereas in mouse acini, the only observed role of mDia was in formation of the F-actin cables perpendicular to the APM, and the TW persisted in the presence of a DN-mDia construct (15). Also in contrast to Drosophila, mouse acini expressing dominant negative (DN)-mDia1 have unchanged basal and stimulated amylase release (15). Clearly, the roles of Dia/mDia1 differ in Drosophila and mouse pancreatic acini.
E. Dynamics of Acinar Actin in the Stimulated State
Actin undergoes a variety of changes during secretory stimulation. The TW in the resting acinar cell is a barrier to zymogen granule (ZG) exocytosis, and the F-actin here must be rearranged to allow ZG access to the APM when the cell is stimulated (26). Using streptolysin O-permeabilized acinar cells, it was demonstrated that mild perturbation of the actin cytoskeleton by adding low concentrations of G-actin sequestering proteins (b-thymosin or a gelsolin fragment; Table 2) resulted in amylase release, especially the early phase of release (26). Interestingly, even when Ca2+ was chelated at low levels using EGTA, mild perturbation of the actin cytoskeleton resulted in amylase release. If high concentrations of the G-actin ABPs were used that resulted in extensive loss of F-actin, there was reduced amylase release. Similarly, if the F-actin stabilizing drug phalloidin was introduced to permeabilized acini, amylase release was inhibited. Together, these data indicate that the TW is a barrier to ZG exocytosis and that, while some remodeling of the actin cytoskeleton is required for release, extensive disruption or stabilization of F-actin is detrimental to protein secretion.
The dynamics of G-actin per se have not been investigated in relation to acinar cell stimulation. However, there have been measurements of the relative changes in the total amount of F-actin in acinar cells, which show that an increase in polymerized actin is associated with amylase release (6). Furthermore, pharmacological reduction of F-actin (1.0 mM latrunculin, a G-actin sequestering agent, Table 2) inhibits CCK-stimulated amylase release from mouse acini without affecting Ca2+ dynamics (7). Since an acute increase in F-actin means less G-actin, these data add to the concept that dynamic changes in actin are involved in protein secretion in the acinar cell.
A similar conclusion regarding actin remodeling during secretion can be drawn from experiments using supramaximal CCK which induces large alterations in the actin cytoskeleton and shows inhibition of amylase release (10, 40). When DN-Rho and DN-Rac (Table 2) constructs were used, they prevented supramaximal CCK inhibition of amylase release, and at the same time lessened reorganization of the actin cytoskeleton (6). In that study a low dose of jasplakinolide (1 mM) also prevented supramaximal CCK inhibition of amylase release, but a higher dose (3 mM) inhibited amylase release at all concentrations of CCK, consistent with the effect of jasplakinolide reported by Valentijn et al (43) (Table 2). So again, these results show there is a 'sweet-spot' for amylase release with respect to organization of the acinar cell actin cytoskeleton.
Remarkably, during exocytosis the ZG membrane (ZGM) becomes 'coated' with F-actin (actin-coated granules, ACG) (43) (Figure 1, right panel). In the acinar cell, the role of the F-actin coat has been proposed to stabilize the fused ZG at the PM (20, 41). This may be related to the duration of acinar protein secretion (seconds, which is long relative to neuronal exocytosis which occurs on a millisecond time scale), which may be required for the highly concentrated digestive enzymes to solubilize and diffuse out of the granule lumen into the beginning of the pancreatic ductal system. In other cells with the regulated pathway whose contents are more rapidly solubilized, the F-actin coat appears to flatten the fused vesicle thus quickly forcing out the secretory product. The potential differences in the roles of F-actin granule/vesicle coats in various secretory cells are discussed in some excellent reviews (28, 29). Interestingly, the G-actin sequestering drug latrunculin blocks coating of ZG with actin but does not prevent ZGM-PM fusion (17).
Using the C3 Rho toxin from Clostridium botulinum, it was shown that ACG formation is Rho-dependent (27). When acini were pretreated with the C3 toxin and then stimulated to secrete, ACG were not observed but large vacuoles formed that were filled with the fluorescent extracellular tracer, showing that granule fusion had still occurred but that the individual granules in the absence of actin coating appear to fuse into a large vacuole. Thus, the rapid formation of ACG in stimulated cells appears to be Rho-dependent, and ACG formation may prevent vacuole formation. Amylase release was not measured in these experiments.
The timing of F-actin coating of ZG relative to ZGM-PM fusion has evolved since its discovery. Early descriptions of ACG suggested that the coat developed before ZGM-PM fusion, based on failure of entry of a fluorescent lectin (WGA) from the medium into ACG, indicating their lumens were not yet accessible to the medium (43). Later experiments concluded that the F-actin coat forms after membrane fusion, while the exocytic pore is still open (27, 41) (also see Pancreapedia article Visualization of exocytosis in pancreatic acinar cells by fluorescence microscopy). This conclusion was based on the observation that only ZG accessible to the acinar lumen (able to take up lysine-fixable fluorescent dextran from the medium) were actin-coated and no ZG without fluorescent dextran were actin-coated. An example of acini stimulated in the presence of a lysine-fixable dextran followed by fixation and fluorescent phalloidin staining is shown in Figure 2. This clearly shows dextran in the lumens of ACG. Subsequent work using a soluble tracer (sulforhodamine B) with living cells convincingly showed that access to the ZG lumen occurred on average 6.7 sec before appearance of the actin coat, as visualized in acini from a transgenic mouse expressing Lifeact-GFP (17) (Figure 3). This study also showed that latrunculin blocked formation of ACG while ZG fusion was not inhibited (Figure 4). Hence, ZGM-PM fusion occurs rapidly followed by actin coating of the fused ZG while it is still in the 'Ω' shape, and that fusion per se is independent of ACG formation.
A different interpretation of the timing of ZG fusion and ACG formation has been made recently by Geron et al., using Lifeact-GFP, expressed in acini by adenoviral infection (15).
Figure 3. Actin coating of ZG occurs shortly after ZGM-PM fusion.[From (17), Fig.4] "Real-time imaging of exocytic fusion events and F-actin coating. (A) Low magnification images show a lumen lying diagonally between two acinar cells identified with SRB (red) and Lifeact-EGFP (green) in the sub-apical region. (A, i) is an image taken before the appearance of exocytic events at the time point indicated ‘‘i’’ on the graph of fluorescence intensity over time in panel (C). (A, v) Is an image at a time point after induction of a number of exocytic events which can be seen as bright spots of SRB fluorescence along the lumen; the time point ‘‘v’’ is indicated on the lower graph (C). (B) Shows an image sequence from an enlarged region (box shown in A) of Lifeact-EGFP and SRB and the overlay, for two exocytic events. The images were taken at the time points i, ii, iii, iv, and v as indicated on the graph in panel (C). (C) is a graph of fluorescence changes over time taken from a region of interest placed over the lower exocytic event (indicated by an arrowhead). SRB fluorescence is plotted normalized to the peak and rises rapidly to a peak and then decays to a plateau. The simultaneously recorded Lifeact-EGFP signal, plotted in arbitrary fluorescence units, rises slowly and nearly reaches a maximum by the end of the record. The starting points of the SRB and Lifeact-EGFP signals, as determined by a positive deflection of the signal by more than 5 times the standard deviation of the signal noise, are shown by the colour-coded triangles on the X axis. The black dotted lines were mono-exponential fits to the data with t values of 6.9 s (SRB) and 29.4 s (Lifeact-EGFP). doi:10.1371/journal.pone.0039815.g004"
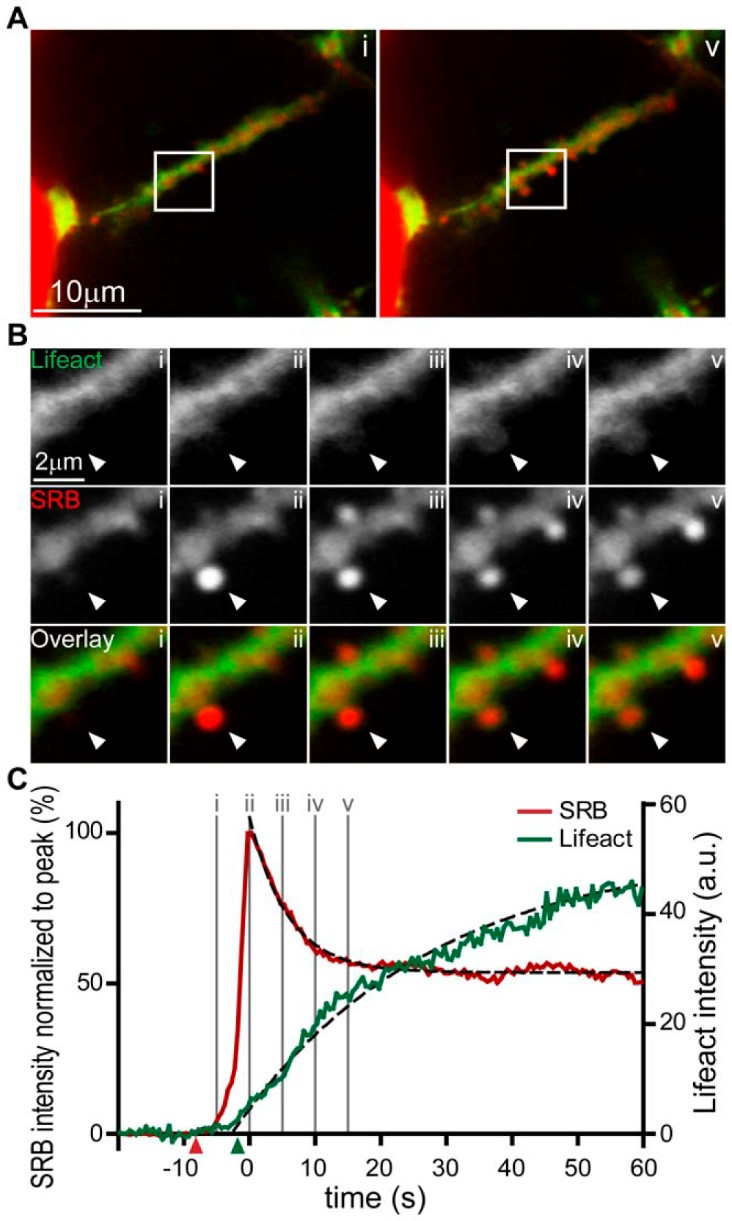
Figure 4. Latrunculin A abolishes actin coating of ZG but does not inhibit ZGM-PM fusion. [From (17), Fig.5] "Latrunculin treatment abolishes F-actin coating of fused granules. (A) Low magnification images show complex lumens, identified by SRB (red) and Lifeact-EGFP fluorescence (green), lying between the cells within a pancreatic fragment. (Ai) is an image taken before the appearance of exocytic events, (Av) is taken after, at the time points ‘‘i’’ and ‘‘v’’ as indicated on the graph in panel (C). (B) Shows an image sequence from an enlarged region (box shown in A) of Lifeact-EGFP and SRB and the overlay, for a single exocytic event. The images were taken at the time points i, ii, iii, iv, and v as indicated on the graph in panel (C). (C) Is a graph of fluorescence changes over time taken from a region of interest placed over the exocytic event (indicated by an arrow). SRB fluorescence is plotted normalized to the first, rapid peak and shows a rapid rise followed by a slower increase. The simultaneously recorded Lifeact-EGFP signal, plotted as arbitrary fluorescence units, shows only very small changes over time. doi:10.1371/journal.pone.0039815.g005"

This study was of the role of the formin family member diaphanous (DIAP1, called mDia in mouse), which is a Rho effector whose activation leads to linear MFs in the pancreatic acinar cell that they called actin cables. They state in the introduction to their paper that 'Secretory vesicles undergo actin coating before fusing to the apical membrane...' (emphasis added) and they cite (27, 28, 43). One can observe in their videos accompanying the paper ACG moving apparently along F-actin cables that run perpendicular to the apical PM. Since these ACG appear deeper inside the cells than the PM, it was concluded that coating preceded ZGM-PM fusion. Even though they included FM4-64, a fluorescent membrane tag that should enter fused ZG and label their membranes, in none of the data shown is FM4-64 fluorescence ever observed in ZG. Therefore, it is not clear that the experiments actually could reveal when ZGM-PM fusion occurred. A more consistent interpretation of the role of mDia-induced actin cables is that fused ZG are moving along these cables near the apical PM.
Another issue with their interpretation is that it does not concur with what was observed when the cables were interfered with. They showed that expression of a CA- mDia1 resulted in an increase in actin cables, and that a DN-mDia1 mutant caused a strong decrease in the number of cables. Neither of these affected basal or stimulated amylase release. If the cables are an important mechanism to move ZG in the cytoplasm to the PM so they can then fuse, one would expect a decrease in stimulated amylase release when the cables are decreased, which was not observed. The DN-mDia1, which reduced cable formation, caused an increase in ACG fused to one another, which was interpreted as compound exocytosis. So, it would appear that the role of these mDia1 induced actin cables is to keep ZG separated such that individual granule fusion predominates and minimizes compound exocytosis.
It is likely an issue in these disparate results regarding the timing of ACG formation whether the acinar preparation was optimal for allowing access from the medium to the acinar lumen. This is discussed in 20, 46), where it is shown that the vigor of the tissue dissociation technique can yield groups of cells ranging from multi-acinar clusters to single acini or even smaller than acinar-sized groups that retain normal acinar secretory function. To allow access of the bathing solution to the acinar lumen, groups consisting of as few as 3-6 cells with a short unhindered connection between the acinar lumen and the medium is optimal. [In my lab, we could not get lysine-fixable dextran into fused ZG (37) as achieved by other labs, likely because my 'acini' were actually larger multi-acini clusters.]
Although mDia is a downstream effector of activated Rho, introduction of DN-mDia (15) and DN-Rho A (6) in acinar cells does not produce the same effects (Table 2). DN-mDia does not affect amylase release whereas DN-Rho inhibits amylase release. These different effects on amylase release indicate that mDia is not the sole target of activated Rho in the acinar cell, and perhaps another of the Rho effectors is involved (39).
The conclusion that the F-actin coat develops rapidly after ZGM-PM fusion contributes to the idea that this coat functions to stabilize the pore in the open state, providing sufficient time for the stored zymogens to become soluble and diffuse into the acinar lumen. An actin coat attached along the cytosolic surface of the fused ZG would stiffen this membrane, preventing closure of the energetically-unfavorable pore. The presence of the nonclassical myosin Vc (Myo5c) on ZG, possibly tethered to the membrane by the small GTPase Rab27B (11, 12) is believed to provide a motive force to move ZG along the TW. Another myosin, Myo2A, is localized to the apical pole of acinar cells, is phosphorylated when the cell is stimulated, and when its activity is inhibited access of the ZG lumen to the soluble markers in the medium is reduced (5). This was interpreted as more rapid fusion pore closure when this myosin activity is inhibited. Also, when the TW was disassembled using cytochalasin treatment, the acinar lumen enlarged under stimulation but amylase was not released; when the cytochalasin and secretory stimulus was washed out, the TW recovered and amylase was released from the already fused ZG (44). It was suggested that contractile forces on the fused ZGM by actin were needed to force the ZG content out. These results can be reinterpreted in light of the newer data indicating that MFs are required to maintain an open pore for protein release.
In addition to providing sufficient time for zymogens to dissociate and diffuse into the lumen, the actin-coat of fused ZG has been proposed to help counteract the expected hydrostatic pressure of these dissolving contents from the fused ZG in salivary glands (22). Also, there is compelling evidence that the fused ZGM transports electrolytes and fluid from the acinar cytosol into the lumen during protein secretion (see review in the Pancreapedia by Frank Thévenod; Channels and Transporters in Zymogen Granule Membranes and their Role in Granule Function: Recent Progress and a Critical Assessment) which will add to hydrostatic pressure within the fused ZG lumen.
While it has been shown that mDia1 is involved in the formation of actin cables in the acinar cell, and is regulated by the small GTPase Rho, it remains to be determined what controls localized disassembly of the TW required to allow passage of ZG to the PM for exocytosis. The F-actin structure that coats granules is likely to consist of a network of short interconnected linear filaments, or alternatively, of branching actin filaments. This is because linear F-actin filaments <10 mm in length are rigid rods (8) and such long linear filaments could not be accommodated on the highly curved surface of granules which are ~1 mm in diameter (34). Consistent with this idea, a subunit of the ARP2/3 complex and N-WASP, factors involved in nucleation of branches on existing MFs, were immunolocalized to the region of ACG in stimulated acini (15). Based on this, Rac1 may be involved, because Rac1 has been shown to regulate WASP/WAVE ABPs that control nucleation/elongation of branched MF in other systems (30). However, WASP nucleates a new branch on existing F-actin and proteomic analysis of highly purified ZGM revealed the presence of small GTPases and molecular motors (dynein, myosins) but not actin or other cytoskeletal elements (11, 12, 33). On the other hand, ultrastructural immunogold localization showed actin in the region of ZG (1, 3). Together these results suggest that actin in the ZG region of the cell is either loosely bound or not bound at all to the ZGM. In any case, it appears F-actin is not present on ZG before fusion, so there needs to be a process to nucleate new MF on the fused ZGM and at this time, how this occurs remains to be identified.
F. After the Stimulus Ends
Once the fused ZG has released its content and exocytosis can be considered complete, the actin cytoskeleton has the further role of helping drive compensatory endocytosis. In order to maintain the balance of membranes between apical and basolateral domains, the ZGM added to the apical surface during protein secretion must be taken back into the cell. The endocytic process is believed to retrieve the ZGM in a clathrin-mediated fashion, and this also requires the actin cytoskeleton (44). It was observed in cytochalasin D treated acinar cells that the APM became dilated and pits on this surface were immunoreactive for the clathrin adaptor AP-2, clathrin, dynamin, and caveolin, all of which are components of various endocytic mechanisms. When cytochalasin was washed out, these proteins rapidly disappeared from the APM and the luminal membrane recovered to its prestimulus size. From this it was suggested that the actin cytoskeleton is also involved in the compensatory endocytosis process that follows protein secretion. The roles of actin in endocytosis are well known (24).
IV. Summary
There are several filamentous actin structures with important roles in acinar cell protein secretion: the TW, ACG, and actin cables. There is also BLPM associated F-actin which is involved in membrane blebbing under supramaximal stimulation. The recent development of the fluorescent Lifeact probe has begun to reveal the dynamics of the actin cytoskeleton with new insights into post-fusion actin coating of ZGs and the discovery of actin cables perpendicular to the TW. It is expected this tool will continue to be utilized for greater in depth understanding of the actin cytoskeleton and its roles in protein secretion from the acinar cell.
V. Addendum
Since this chapter was written in 2015, a PubMed search was carried out in October 2020 and showed that recent work concerning the actin cytoskeleton in acinar cells was primarily focused on endocytosis and autophagy and not digestive enzyme secretion. By contrast, recent work exists on actin in islet beta cells and its relationship to insulin secretion. This addendum will review this recent work on the acinar cell actin cytoskeleton in endocytosis linked to autophagy during models of pancreatitis and in insulin release from islet β cells.
A. Role of Actin in Acinar Cell Autophagy and Pancreatitis
Autophagy is a process whereby cells breakdown and recycle their own components (A7). Autophagy can be induced by starvation thus providing the cell with energy and substrates to survive this stress. Additionally, and relevant to this discussion, cytotoxic events in the cell can also induce autophagy in an attempt to clear malformed, cytotoxic organelles and materials, again to save the cell and/or protect the wider organism. In the canonical autophagy pathway, this occurs by formation of an autophagosome, a double membrane structure, to engulf the damaged organelle/material which then fuses with a lysosome to degrade it. In the non-canonical form of autophagy, the machinery that drives phagosome formation (a marker of which is LC3, microtubule-associated protein 1A/1B-light chain 3) is recruited to a single membrane organelle which promotes their fusion with lysosomes for degradation of the organelle contents.
During pancreatitis, there is widespread disruption of ongoing protein synthesis and the packaging of zymogens into functional secretory granules (for review see (A12)). For example, during alcohol-induced pancreatitis, zymogen synthesis is increased at the transcriptional level and, at the same time, exocytosis is inhibited resulting in an overabundance of zymogen granules in the cell. Because of the high synthetic rate, the capacity of the protein folding machinery of the endoplasmic reticulum (ER) is exceeded leading to an unfolded protein response by the cell. These conditions promote activation of zymogens, especially important of which is activation of trypsin from trypsinogen. Active trypsin in turn can lead to cell/organ damage and is accompanied by inflammation which together enhance the severity of pancreatitis.
One of the cell responses to stress, as mentioned above, is to activate autophagy, in this case to get rid of the excess of zymogen granules. In pancreatitis, there is a strong body of evidence that lysosomal biogenesis fails to keep up and that there is insufficient flux into the autophagy pathway such that trypsin activation occurs in the cell with its damaging sequelae.
As with all membrane trafficking, formation and movement in the autophagy pathway involves cytoskeletal elements, including actin (see (A6) for review). Evidence for this includes the observation that actin-depolymerizing agents (e.g., cytochalasin D, latrunculin B) block autophagosome formation. The actin regulating Rho family GTPases are involved also. RhoA is needed during starvation induced autophagy, while Rac is inhibitory to induction and itself is inactivated during autophagy. The GTPase Rab1 is also involved, stimulating actin assembly and tubulation of the ER membrane to produce autophagosomes.
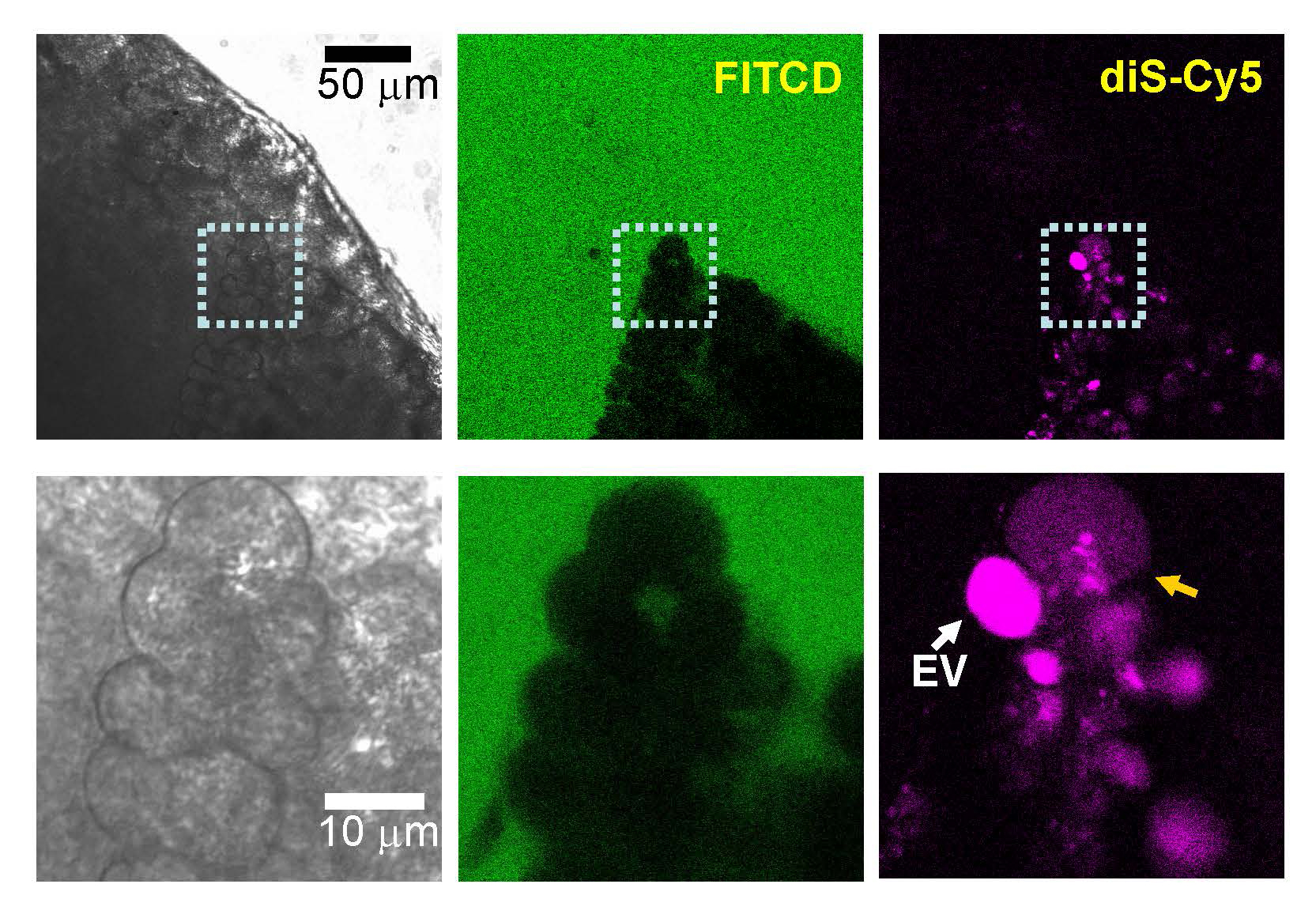
Recent studies investigated the role of endocytosis, which is well known to involve actin, in experimental pancreatitis. Chvanov et al. used LifeAct to follow actin dynamics during pancreatitis along with extracellular fluorescent tracers to monitor fluid phase endocytosis (A1). They found that endocytic structures that form during pancreatitis are much larger than during physiological stimulation, forming endocytic vacuoles (EV) which are positive for active trypsin. These EVs can be as large as 12 μm in diameter and appear to form from the enlarged apical plasma membrane that result from compound exocytosis during acinar protein release. These EVs are actin-coated, often not continuously. The small ‘fenestrae’ in the actin coat that were sometimes observed appear to be weak spots where rupture of EVs can occur (as revealed by movement of the fluid phase fluorescent tracer from the EV to the cytosol; see Figure 5, reproduced from this paper) and deliver active trypsin to the cytosol. Once rupture occurs it is quickly followed by plasma membrane blebbing in most of their observations and cell death about 30 min later. Importantly, the presence of the F-actin stabilizing drug jasplakinolide decreased the rupture of EVs from about 30% of observed cells down to about 15%. This led the authors to suggest that the actin coat on the EVs is a stabilizing force against rupture and is cell protective.
Figure 5. Cytosolic presence of membrane-impermeant fluorescence probe in the cell located in undissociated pancreatic fragment. [From (A1); with permission of the author, A.V.Tepikin]. “Small (~1 mm) section of mouse pancreas was stimulated by 100 nM CCK for 2 h at 35°C in the presence of diS-Cy5 (shown in magenta), washed and imaged in the presence of FITCD (shown in green). The lower gallery of images depicts the fragment containing two cells within the section: one with a large intact EV (white arrow) and the adjacent cell with increased cytosolic fluorescence of diS-Cy5. The FITCD image indicates that the plasma membrane of this cell is intact, suggesting that the increase of the cytosolic fluorescence occurred as a result of EV rupture. Representative of six similar experiments.”
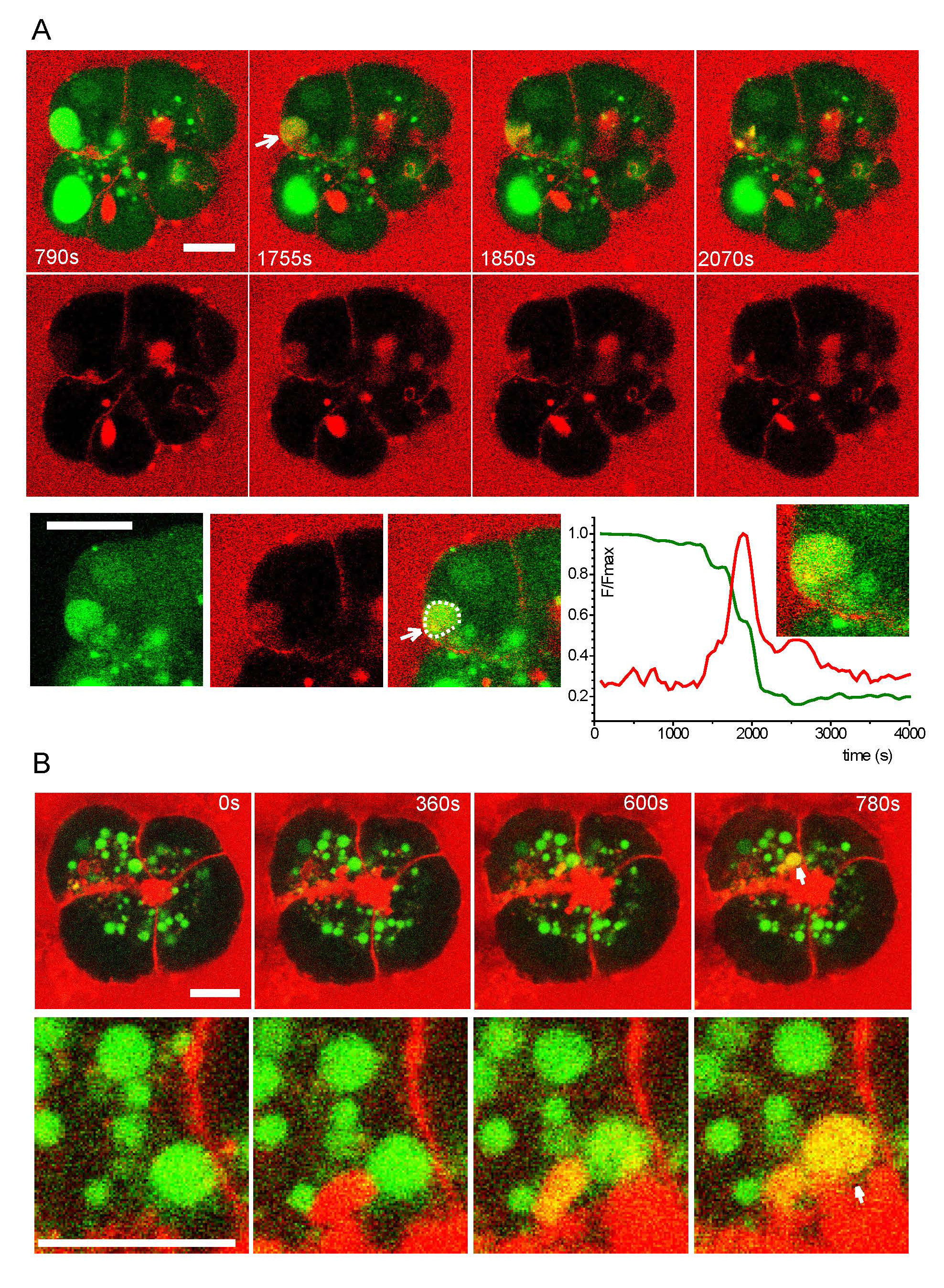
Another interesting observation was that EVs can sometimes fuse with the plasma membrane, as revealed by mixing of preloaded fluorescent tracer of one color with a differently colored tracer in the medium (A1). This exocytosis of EVs occurred at both the apical and basolateral surfaces of the cells (see Figure. 6, reproduced from (A1). Because of the technical setup for these high resolution confocal studies, not many individual events could be recorded so it is not known how frequently this occurs or whether the propensity is for apical or basolateral exocytosis. The basolateral exocytosis is consistent with that investigated by Gaisano and colleagues (A3) which is proposed as mechanism to deliver harmful active trypsin to the interstitial space during pancreatitis. The process of exocytosis from the autophagic pathway has been termed secretory autophagy (see (A4) for review).
Figure 6. Exocytosis of endocytic vacuoles (EVs).
[From (A1); with permission of the author, A.V.Tepikin]. “A, fusion of LY-filled EV (green; the EV undergoing fusion is highlighted by an arrow) with the basal plasma membrane of a CCK-stimulated acinar cell. The extracellular medium contains TRD (red) and a fusion event is characterized by mixing of the probes (yellow). Following fusion, the EV loses LY and gains TRD. The region containing the fusing EV is shown in the bottom row of images. The graph shows the time-course of fluorescence of the two dyes recorded in the EV (highlighted by the dashed line). B, LY-filled EV (green) fuses with TRD-filled (red) post-exocytic structure in the apical region of the CCK-stimulated cell. The region containing the fusing EV (white arrow) is shown in the bottom row of images. Following fusion, the composite structure contains both probes (yellow) (representative of n = 7 observations). Scale bars = 10 μm.”
Subsequent work from this group looked in greater detail at the formation of EVs and presented evidence that they form by a non-canonical pathway, LC3-associated phagocytosis (LAP) (A2). This process involves an EV with a single membrane which is initially actin coated and as it loses actin becomes LC3 positive and is converted into an autophagic organelle. Importantly, most LC3-positive EVs also exhibited activation of trypsinogen.
A different study implicating the actin cytoskeleton in autophagy and pancreatitis studied β1 syntrophin, which binds and regulates dystrophin and is in turn an important linker between the actin cytoskeleton and transmembrane glycoproteins (A15). In this case, actin remodeling driven by β1 syntrophin is involved in curvature of the membrane at the ER to form tubules which become autophagosomes. Using a β1 syntrophin knockout mouse, they observed dilation of the acinar lumen under resting conditions, and the knockout showed an unaltered cerulein dose-response amylase release. However, during cerulein supramaximal stimulation to induce pancreatitis, the β1 syntrophin mice had less F-actin and reduced autophagy: a marked reduction in ER nucleations, autophagosomes, and autolysosomes. The β1 syntrophin knockouts exhibited greater severity of pancreatitis (higher serum amylase and trypsin; more necrosis; more neutrophil infiltration; and greater edema) showing the protective role of autophagy during pancreatitis. At this time, there does not appear to be enough information to know the relative importance of canonical (ER-derived) vs non-canonical (endocytic organelle) autophagic pathways in pancreatic function and in pancreatitis.
B. The Role of Actin in β Cell Exocytosis
Although there are known differences in the cell biology of endocrine and exocrine pancreatic cells, there are more recent investigations of the actin cytoskeleton in the β cell than in the acinar cell. The Pancreapedia, The Exocrine Pancreas Knowledge Base, includes a review of the physiology and cell biology of insulin release and the intracellular pathways controlling it (A9). The final signaling step in β cell exocytosis of insulin granules, similar to most secretory cells, is an influx of Ca2+ resulting in activation of the SNARE complex (soluble N-ethylmaleimide sensitive factor attachment protein receptor). The complex consists of SNAREs on the granule and plasma membranes whose conformational changes provide the force to fuse these two membranes and release the granule content to the extracellular space (A14). The major physiological stimulus for β cell exocytosis is elevated concentration of blood nutrients, especially glucose. Glucose-stimulated insulin release proceeds in two phases (A5, A14) The initial phase results in a large spike of insulin release and lasts about 10min. If elevated glucose is maintained, the second phase occurs, and this is a lower rate of release but is capable of being sustained for hours, ensuring the circulating insulin remains sufficient throughout nutrient uptake from the intestines to assimilate those nutrients by target organs throughout the body.
As in the acinar cell, glucose-stimulated insulin release from the β cell involves remodeling of the actin cytoskeleton, which in the case of the β cell has roles in both phases of exocytosis (see Kalwat and Thurmond for review (A5)). As in other secretory cells, the β cell has a cortical F-actin cytoskeleton which presents a barrier to granule exocytosis, as revealed by actin depolymerizing agents that increase insulin secretion. Actin remodeling is also involved in recruitment of new granules to the plasma membrane during the second, sustained phase of insulin release.
If we look at upstream events, the influx of glucose has two pathways that lead to insulin release (A11, A14). Glucose is taken up from the circulation via GLUT glucose channels in the plasma membrane into the cytosol. Metabolism of glucose in the cell increases the ATP/ADP ratio which inactivates the ATP-sensitive K+ channel resulting in membrane depolarization and an influx of Ca2+, stimulating SNARE-mediated membrane fusion. The other pathway is that elevated glucose leads to glucosylation of Cdc42, a Rho GTPase, and its glucosylation decreases the level of Cdc42-GTP, which is the active form. Activated Cdc42 interacts with two proteins that affect actin remodeling. One is WASP (Wiskott-Aldrich syndrome protein), which through ARP2/3 (actin-related protein complex 2/3), stimulates branching of F-actin. The other is mDia (discussed in main Chapter text), which promotes actin polymerization. Thus, inhibition of Cdc42 results in actin remodeling and increases insulin release in a similar fashion as do actin depolymerizing drugs. Further evidence of the importance of Cdc42 is that a constitutively active form of the protein (Q61L) inhibits glucose-stimulated insulin release; and a constitutively inactive dominant negative form (T17N) is without effect (A11). The authors also used jasplakinolide (nucleates and stabilizes F-actin) which unexpectedly potentiated glucose-stimulated insulin release, without effect on unstimulated (basal) release. Jasplakinolide did not enhance release with KCl depolarization even though the abundance of granules near the plasma membrane was increased. Importantly, glucose stimulation dramatically diminished cortical F-actin whereas KCl depolarization did not. These results indicate that cortical actin is decreased at the same time deeper F-actin is increased during glucose stimulation, thus promoting the initial phase of release and recruiting new granules for the sustained second phase.
They next investigated whether the SNARE syntaxin 1 (plasma membrane localized), which is known to interact with actin, is involved. They immunoprecipitated (IP) syntaxin 1 and measured co-IP actin. Glucose stimulation decreased actin associated with syntaxin 1 ~2-fold. The additional presence of jasplakinolide during glucose stimulation did not affect this decrease in co-IPed actin. Depolarization with KCl had no effect on co-IP of actin, alone or in the presence of jasplakinolide. Therefore, glucose-stimulated actin remodeling occurs even in the presence of the actin nucleating and polymerization effects of this drug. The authors proposed that glucose initiates the transient reduction of filamentous cortical actin to coordinate granule mobilization and pool refilling and thereby promotes granule exocytosis.
Another recent study has investigate F-actin coating of insulin granules with a focus on ARP2/3, a nucleator that leads to branched F-actin filaments (A8). Using endocrine pancreas cells from LifeAct transgenic mice and a fluorescent fluid-phase endocytic marker, with multiphoton microscopy, they provide high resolution data showing that insulin granules become actin coated following membrane fusion (labeling with the fluid-phase marker preceded actin coating) and that this is ARP2/3 dependent. Using drugs that block ARP2/3 activity and short hairpin RNA (shRNA) to deplete the cells of the protein, they observed loss of F-actin coating. Interestingly, with this high resolution technique they found that there was not a global change in the cortical F-actin cytoskeleton but only localized changes to individual fusing granules. When ARP2/3 was inhibited or absent, fused granules accumulated at the plasma membrane and insulin release was decreased even though fusion had occurred. They then examined whether linear F-actin has a role by interfering with formin and find that while actin coating was disrupted, insulin release was unaffected. The authors concluded that ARP2/3-mediated nucleation of branched F-actin plays an active role post-fusion in events involved in insulin release.
Because of the complexity, especially delineating the two phases of insulin release and their regulatory mechanisms, a recent study presented the initial development of a statistically-based computer model along the lines of Cellular Autamoton, to integrate what is known and provide a platform for evaluating new data and hypotheses regarding the role of the actin cytoskeleton in insulin granule transport in the β cell (A10). Since the model they develop needs to incorporate what is known about β cell insulin release, this paper also serves as an up to date review of granule movement and the various roles of the actin cytoskeleton in the β cell.
The authors interpret recent TIRF (total internal-reflection fluorescence: a very narrowly focused signal with high signal to noise, in close proximity to the plasma membrane that reveals only granules docked/primed and can reveal exocytic events), and confocal data that insulin granules can be docked at the plasma membrane (responsible for first rapid phase of insulin release); can be freely diffusible toward the plasma membrane and back again deeper into the cytosol; and can be anchored via various accessory proteins to F-actin filaments, such that these are being transported and will be release during the prolonged second phase.
They set up their initial model such that “the following processes and hypotheses are considered:
● Granules are created inside the cell.
● Granules move either diffusely or are directed, with stimuli promoting a directed movement.
● Granules can move along actin fibers, and are prevented from moving perpendicular to the strand.
● With a certain probability, granules can pass through the actin network, in cases of actin remodeling.
● Granules must exist for a certain time before they can fuse at the membrane.
● Under constant stimuli, a steady state exists for insulin secretion.”
In the simulation, they reduce a β cell to a cube 10 μm on each edge with 343,000 cubic elements approximating the size of a single insulin granule. Focusing on just granules and actin, they define 11 possible states for each element denoting granules in various associations with actin, its movement and the granule’s age (new or in the immediately releasable pool). With this model they can investigate the actin cytoskeleton and how it relates to granule movements and insulin release. To set up initial conditions they used fluorescence microscopy data from LifeAct expressing cultured mouse islets and MIN6 cells. The simulation is populated with actin filaments and granules, randomly assigned to fit the known data. For changes in granule behavior (movement, actin-association, fusion, etc.) the stimulus (glucose, KCl depolarization) and intracellular Ca2+ levels are used to define the probabilities of such changes. They start the simulation by randomly placing 13,000 granules within the actin mesh and assigning the majority to diffusing and a small fraction to different motions, to match data in the literature regarding these parameters.
The simulations were run and compared visually to see how well the simulations approximate actual cells, and then quantitatively to see how stimuli affect insulin release. They also used latrunculin to disassemble F-actin and compared results from real cells with the simulations.
In summary, the authors conclude that “three major results were achieved:
1. The simulated pattern of insulin secretion in response to high glucose shows the typical biphasic pattern of cultured islets and cell lines
2. The simulated effect of the modifier of the actin cytoskeleton, Latrunculin B, namely the increase of both phases of glucose-induced secretion, concurs with experimental observations, provided the simulated network fulfils a certain set of criteria.
3. This network is also compatible with a simulated interaction between glucose- and potassium-stimulated secretion, which mirrors earlier experimental observations.”
“…Thus, on the one hand, extensions can be implemented informatically in an uncomplicated way, and on the other hand, computing capacities are not yet a problem. Rather, the addition of further findings and hypotheses is an important component in the further development of the model, so that quantitative predictions should also be possible in the future.”
In conclusion, although there has been little recent progress on investigating the roles of the actin cytoskeleton in acinar cell protein secretion under normal physiological conditions, there are new data in the context of pancreatitis as well as in other systems such as the β cell. The phasic release of insulin from the β cell shows several roles for actin and its remodeling. It should be emphasized that much of this progress relied on use of high resolution live cell imaging. The TIRF signal is limited to ~100 nm from the coverslip surface into the attached cell. Similarly, multiphoton microscopy is able to focus into approximately 600 nm thick focal plane of interest. These techniques exclude signals outside of these thin focal planes, thus considerably reducing background that would obscure the region of interest. Interestingly, there are also kinetic differences for zymogen secretion from granules that are older or newly synthesized (e.g., see (A13)). The issue of release of old vs new zymogen granules has not been explored with respect to actin, but it would seem likely to be important to this process.
VI. References
- Becich MJ, Bendayan M and Reddy JK. Intracellular transport and storage of secretory proteins in relation to cytodifferentiation in neoplastic pancreatic acinar cells. J Cell Biol 96: 949-960, 1983. PMID: 6833397.
- Belyantseve IA, Perrin BJ, Sonnemann KJ, Zhu M, Stepanyan R, McGee J, et al. Gamma-actin is required for cytoskeletal maintenance but not development. Proc Natl Acad Sci 106: 9703-9708, 2009. PMID: 19497859.
- Bendayan M. Ultrastructural localization of cytoskeletal proteins in pancreatic secretory cells. Can J Biochem Cell Biol 63: 680-690, 1985. PMID: 2412674.
- Bendayan M, Marceau N, Beaudoin AR and Trifaro JM. Immunocytochemical localization of actin in the pancreatic exocrine cell. J Histochem Cytochem 30: 1075-1078, 1982. PMID: 7130670.
- Bhat P and Thorn P. Myosin 2 maintains an open exocytic fusion pore in secretory epithelial cells. Mol Biol Cell 20: 1795-1803, 2009. PMID: 19158378.
- Bi Y, Page SL and Williams JA. Rho and Rac promote acinar morphological changes, actin reorganization, and amylase secretion. Am J Physiol Gastrointest Liver Physiol 289: G561-G570, 2005. PMID: 15920016.
- Bi Y and Williams JA. A role for Rho and Rac in secretagogue-induced amylase release by pancreatic acini. Am J Physiol Cell Physiol 289: C22-C32, 2005. PMID: 15743890.
- Blanchoin L, Boujemaa-Paterski R, Sykes C and Plastino J. Actin dynamics, architecture, and mechanics in cell motility. Physiol Rev 94: 235-263, 2014. PMID: 24382887.
- Brown JW and McKnight CJ. Molecular model of the microvillar cytoskeleton and organization of the brush border. PLoS ONE 5: e9406, 2010. PMID: 20195380.
- Burnham DB and Williams JA. Effects of high concentrations of secretagogues on the morphology and secretory activity of the pancreas: a role for microfilaments. Cell Tissue Res 222: 201-212, 1982. PMID: 6174234.
- Chen X, Li C, Izumi T, Ernst SA, Andrews PC and Williams JA. Rab27b localizes to zymogen granules and regulates pancreatic acinar exocytosis. Biochem Biophys Res Commun 323: 1157-1162, 2004. PMID: 15451418.
- Chen X, Walker AK, Strahler JR, Simon ES, Tomanicek-Volk SL, Nelson BB, et al. Organellar proteomics: Analysis of pancreatic zymogen granule membranes. Mol Cell Proteomics 2005. PMID: 16278343.
- Clark AG, Dierkes K and Paluch EK. Monitoring actin cortex thickness in live cells. Biophys J 105: 570-580, 2013. PMID: 23931305.
- Deibler M, Spatz JP and Kemkemer R. Actin fusion proteins alter the dynamics of mechanically induced cytoskeleton rearrangement. PLoS ONE 6: e22941, 2011. PMID: 21850245.
- Geron E, Schejter ED and Shilo BZ. Directing exocrine secretory vesicles to the apical membrane by actin cables generated by the formin mDia1. Proc Natl Acad Sci U S A 110: 10652-10657, 2013. PMID: 23754409.
- Glenney JR, Jr., Glenney P and Weber K. The spectrin-related molecule, TW-260/240, cross-links the actin bundles of the microvillus rootlets in the brush borders of intestinal epithelial cells. J Cell Biol 96: 1491-1496, 1983. PMID: 6841456.
- Jang Y, Soekmadji C, Mitchell JM, Thomas WG and Thorn P. Real-time measurement of F-actin remodelling during exocytosis using Lifeact-EGFP transgenic animals. PLoS ONE 7: e39815, 2012. PMID: 22768313.
- Kaur S, Norkina O, Ziemer D, Samuelson LC and De Lisle RC. Acidic duodenal pH alters gene expression in the cystic fibrosis mouse pancreas. Am J Physiol Gastrointest Liver Physiol 286: G480-G490, 2004. PMID: 15064229.
- Kloepper TH, Kienle CN and Fasshauer D. An Elaborate Classification of SNARE Proteins Sheds Light on the Conservation of the Eukaryotic Endomembrane System. Mol Biol Cell 18: 3463-3471, 2007. PMID: 17596510.
- Larina O, Bhat P, Pickett JA, Launikonis BS, Shah A, Kruger WA, et al. Dynamic regulation of the large exocytotic fusion pore in pancreatic acinar cells. Mol Biol Cell 18: 3502-3511, 2007. PMID: 17596517.
- Mannherz HG, Goody RS, Konrad M and Nowak E. The interaction of bovine pancreatic deoxyribonuclease I and skeletal muscle actin. Eur J Biochem 104: 367-379, 1980. PMID: 6244947.
- Masedunskas A, Sramkova M and Weigert R. Homeostasis of the apical plasma membrane during regulated exocytosis in the salivary glands of live rodents. Bioarchitecture 1: 225-229, 2011. PMID: 22754613.
- Massarwa R, Schejter ED and Shilo BZ. Apical secretion in epithelial tubes of the Drosophila embryo is directed by the Formin-family protein Diaphanous. Dev Cell 16: 877-888, 2009. PMID: 19531358.
- Mooren OL, Galletta BJ and Cooper JA. Roles for actin assembly in endocytosis. Annu Rev Biochem 81: 661-686, 2012. PMID: 22663081.
- Morrison SS and Dawson JF. A high-throughput assay shows that DNase-I binds actin monomers and polymers with similar affinity. Anal Biochem 364: 159-164, 2007. PMID: 17397792.
- Muallem S, Kwiatkowska K, Xu X and Yin HL. Actin filament disassembly is a sufficient final trigger for exocytosis in nonexcitable cells. J Cell Biol 128: 589-598, 1995. PMID: 7860632.
- Nemoto T, Kojima T, Oshima A, Bito H and Kasai H. Stabilization of exocytosis by dynamic F-actin coating of zymogen granules in pancreatic acini. J Biol Chem 279: 37544-37550, 2004. PMID: 15184362.
- Nightingale TD, Cutler DF and Cramer LP. Actin coats and rings promote regulated exocytosis. Trends Cell Biol 22: 329-337, 2012. PMID: 22543050.
- Porat-Shliom N, Milberg O, Masedunskas A and Weigert R. Multiple roles for the actin cytoskeleton during regulated exocytosis. Cell Mol Life Sci 70: 2099-2121, 2013. PMID: 22986507.
- Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol 16: 522-529, 2006. PMID: 16949823.
- Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, et al. Lifeact: a versatile marker to visualize F-actin. Nat Methods 5: 605-607, 2008. PMID: 18536722.
- Riedl J, Flynn KC, Raducanu A, Gartner F, Beck G, Bosl M, et al. Lifeact mice for studying F-actin dynamics. Nat Methods 7: 168-169, 2010. PMID: 20195247.
- Rindler MJ, Xu CF, Gumper I, Smith NN and Neubert TA. Proteomic Analysis of Pancreatic Zymogen Granules: Identification of New Granule Proteins. J Proteome Res 2007. PMID: 17583932.
- Risca VI, Wang EB, Chaudhuri O, Chia JJ, Geissler PL and Fletcher DA. Actin filament curvature biases branching direction. Proc Natl Acad Sci U S A 109: 2913-2918, 2012. PMID: 22308368.
- Sabbatini ME, Bi Y, Ji B, Ernst SA and Williams JA. CCK activates RhoA and Rac1 differentially through Galpha13 and Galphaq in mouse pancreatic acini. Am J Physiol Cell Physiol 298: C592-C601, 2010. PMID: 19940064.
- Shin DM, Zhao XS, Zeng W, Mozhayeva M and Muallem S. The mammalian Sec6/8 complex interacts with Ca(2+) signaling complexes and regulates their activity. J Cell Biol 150: 1101-1112, 2000. PMID: 10973998.
- Tandon C and De Lisle RC. Apactin is involved in remodeling of the actin cytoskeleton during regulated exocytosis. Eur J Cell Biol 83: 79-89, 2004. PMID: 15146979.
- Theriot JA. Accelerating on a treadmill: ADF/cofilin promotes rapid actin filament turnover in the dynamic cytoskeleton. J Cell Biol 136: 1165-1168, 1997. PMID: 9087434.
- Thumkeo D, Watanabe S and Narumiya S. Physiological roles of Rho and Rho effectors in mammals. Eur J Cell Biol 92: 303-315, 2013. PMID: 24183240.
- Torgerson RR and McNiven MA. The actin-myosin cytoskeleton mediates reversible agonist-induced membrane blebbing. J Cell Sci 111 ( Pt 19): 2911-2922, 1998. PMID: 9730983.
- Turvey MR and Thorn P. Lysine-fixable dye tracing of exocytosis shows F-actin coating is a step that follows granule fusion in pancreatic acinar cells. Pflugers Arch 448: 552-555, 2004. PMID: 15103465.
- Uribe R and Jay D. A review of actin binding proteins: new perspectives. Mol Biol Rep 36: 121-125, 2009. PMID: 17939058.
- Valentijn JA, Valentijn K, Pastore LM and Jamieson JD. Actin coating of secretory granules during regulated exocytosis correlates with the release of rab3D. Proc Natl Acad Sci U S A 97: 1091-1095, 2000. PMID: 10655489.
- Valentijn KM, Gumkowski FD and Jamieson JD. The subapical actin cytoskeleton regulates secretion and membrane retrieval in pancreatic acinar cells. J Cell Sci 112: 81-96, 1999. PMID: 9841906.
- Williams JA. Effects of cytochalasin B on pancreatic acinar cell structure and secretion. Cell Tissue Res 179: 453-466, 1977. PMID: 862010.
- Williams, JA. Isolation of rodent pancreatic acinar cells and acini by collagenase digestion. Pancreapedia: Exocrine Pancreas Knowledge Base, 2010. DOI: 10.3998/panc.2010.18.
- Xue B and Robinson RC. Guardians of the actin monomer. Eur J Cell Biol 92: 316-332, 2013. PMID: 24268205.
- Yao X, Chaponnier C, Gabbiani G and Forte JG. Polarized distribution of actin isoforms in gastric parietal cells. Mol Biol Cell 6: 541-557, 1995. PMID: 7663022.
VII. Addendum's References
A1. Chvanov M, De Faveri F, Moore D, Sherwood MW, Awais M, Voronina S, Sutton R, Criddle DN, Haynes L, and Tepikin AV. Intracellular rupture, exocytosis and actin interaction of endocytic vacuoles in pancreatic acinar cells: initiating events in acute pancreatitis. J Physiol 596: 2547-2564, 2018. PMID: 29717784.
A2. De Faveri F, Chvanov M, Voronina S, Moore D, Pollock L, Haynes L, Awais M, Beckett AJ, Mayer U, Sutton R, Criddle DN, Prior IA, Wileman T, and Tepikin AV. LAP-like non-canonical autophagy and evolution of endocytic vacuoles in pancreatic acinar cells. Autophagy 16: 1314-1331, 2020. PMID: 31651224.
A3. Dolai S, Liang T, Orabi AI, Holmyard D, Xie L, Greitzer-Antes D, Kang Y, Xie H, Javed TA, Lam PP, Rubin DC, Thorn P, and Gaisano HY. Pancreatitis-Induced Depletion of Syntaxin 2 Promotes Autophagy and Increases Basolateral Exocytosis. Gastroenterology 154: 1805-1821 e1805, 2018. PMID: 29360461.
A4. Gonzalez CD, Resnik R, and Vaccaro MI. Secretory Autophagy and Its Relevance in Metabolic and Degenerative Disease. Front Endocrinol (Lausanne) 11: 266, 2020. PMID: 32477265.
A5. Kalwat MA, and Thurmond DC. Signaling mechanisms of glucose-induced F-actin remodeling in pancreatic islet beta cells. Exp Mol Med 45: e37, 2013. PMID: 23969997.
A6. Kast DJ, and Dominguez R. The Cytoskeleton-Autophagy Connection. Curr Biol 27: R318-R326, 2017. PMID: 28441569.
A7. Kawabata T, and Yoshimori T. Autophagosome biogenesis and human health. Cell Discov 6: 33, 2020. PMID: 32528724.
A8. Ma W, Chang J, Tong J, Ho U, Yau B, Kebede MA, and Thorn P. Arp2/3 nucleates F-actin coating of fusing insulin granules in pancreatic beta cells to control insulin secretion. J Cell Sci 133: 2020. PMID: 32079655.
A9. Mann E, and Bellin MD. Secretion of Insulin in Response to Diet and Hormones. In: Pancreapedia: Exocrine Pancreas Knowledge Base. 2015. DOI: 10.3998/panc.2016.3.
A10. Muller M, Glombek M, Powitz J, Bruning D, and Rustenbeck I. A Cellular Automaton Model as a First Model-Based Assessment of Interacting Mechanisms for Insulin Granule Transport in Beta Cells. Cells 9: 2020. PMID: 32570905.
A11. Nevins AK, and Thurmond DC. Glucose regulates the cortical actin network through modulation of Cdc42 cycling to stimulate insulin secretion. Am J Physiol Cell Physiol 285: C698-710, 2003. PMID: 12760905.
A12. Rasineni K, Srinivasan MP, Balamurugan AN, Kaphalia BS, Wang S, Ding WX, Pandol SJ, Lugea A, Simon L, Molina PE, Gao P, Casey CA, Osna NA, and Kharbanda KK. Recent Advances in Understanding the Complexity of Alcohol-Induced Pancreatic Dysfunction and Pancreatitis Development. Biomolecules 10: 2020. PMID: 32349207.
A13. Singh M. Nonparallel transport of exportable proteins in rat pancreas in vitro. Can J Physiol Pharmacol 60: 597-603, 1982. PMID: 6179588.
A14. Thurmond DC, and Gaisano HY. Recent Insights into Beta-cell Exocytosis in Type 2 Diabetes. J Mol Biol 432: 1310-1325, 2020. PMID: 31863749.
A15. Ye R, Onodera T, Blanchard PG, Kusminski CM, Esser V, Brekken RA, and Scherer PE. beta1 Syntrophin Supports Autophagy Initiation and Protects against Cerulein-Induced Acute Pancreatitis. Am J Pathol 189: 813-825, 2019. PMID: 30653956.