
Methods Type:
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2011.15
| Attachment | Size |
|---|---|
| 194.38 KB |
The control of regulated exocytosis is still largely unknown in epithelial cells (12). While much progress has been made in understanding constitutive secretion we still know only in outline the mechanisms of secretory granule fusion (5). Progress requires methods of real-time imaging of granule exocytosis and in recent years a variety of optical methods have been applied towards understanding epithelial cell secretion. The results obtained have ranged from simply counting the numbers of fused granules to insights into the molecular mechanisms that regulate fusion and fission.
Differential interference contrast (DIC) microscopy detects optical changes due to the loss of granule content (3, 4). Its advantages include the ability to record from an unstained preparation. However, it is not possible to track granule behaviour either before or after loss of granule content (10). So although the method has utility in counting fusion events it is not useful for the study of granule behaviour.
In contrast, the fluorescence methods of confocal and two-photon microscopy have been used to study post-fusion granule behaviour.
Confocal imaging of extracellular fluorescent dyes (lucifer yellow) was first used to record exocytosis by Segawa (9). At the point of granule fusion the opening of the aqueous fusion pore allows entry of the fluorescent dye into the granule which is then recorded as a fluorescent change. The major obstacle to using this method in living cells is that the signal is very small compared to the fluorescence noise from the surrounding out of focus fluorescent dye (6). However, in an adaptation of the method, confocal microscopy has found great utility in paraformaldehyde fixed preparations (11). Here lysine fixable extracellular dyes are applied to living tissues. Exocytosis is stimulated and the dye enters the fused granules. Fixation then cross-links the dye in the granules and lumen and subsequent washing of the tissue removes the surrounding dye. The method has been used to show that the zymogen granule fusion pore is capable of opening and closing (7). It is also a method that can be combined with counter immunohistochemistry to determine the involvement of proteins like F-actin and myosin 2A in exocytosis in the secretory process (2).
For live-cell imaging two-photon microscopy gets around the problems of signal to noise in confocal imaging. Two photon microscopy has been used to reveal the nature of compound exocytosis (8) and the kinetics of zymogen granule fusion (7) in pancreatic acinar cells.
This methods article focuses on a detailed description of the methods used in confocal microscopy of fixed exocrine tissue and live-cell two photon microscopy.
1. Materials
- Pancreases are excised from mice around 6 weeks old (~25g).
- Na-rich extracellular buffer contains (mM): NaCl 145, KCl 5, CaCl2 2, MgCl2 1, Glucose 10, HEPES 10, pH 7.4.
- Worthington CLSPA collagenase is dissolved into extracellular Na-rich buffer to a final concentration of 250 units per ml.
- The lysine fixable extracellular dyes we use include both 3000 and 10,000 MW. For example we routinely use FITC-dextran (Invitrogen D3306, D1820) and TMRE-dextran (Invitrogen D3308). The dextran dyes are used at a final concentration of 2 mg/ml. We often use fluorescent phalloidins to stain the apical F-actin web (eg Rhodamine phalloidin, Invitrogen R415) with a final dilution to a concentration of 5 units/mL.
- Glass cover slips are coated with poly-l-lysine (Sigma-Aldrich P8920 0.1% solution w/v) by applying a drop of poly-l-lysine solution to the cover slip for 5 minutes and then removing and drying.
- Centrifuges, water baths etc are all standard laboratory equipment.
- We have used Zeiss LSM510 and Olympus FV1000 confocal microscopes and for two photon microscopy we have used a Leica TCS SP2 and laboratory-built machines.
2. Methods
2.1 Preparation of pancreatic fragments
The aim of this method is to produce fragments of pancreatic exocrine tissue that span a range of sizes from around 10 cells up to about 200 acinar cells. The smaller fragments retain apical-basal morphology and have remnants of the lumen. The small fragments are readily permeabilized and are ideal for antibody penetration in immunohisotchemistry experiments. In contrast the larger fragments retain lengths of lumens often branching and extending to over 100 mm long. Most show evidence of terminating lumens indicative that they are secretory end-pieces. These larger tissue fragments are ideal for live-cell two photon imaging.
In our protocol we inject 750 ml of CLSPA collagenase in Na-rich extracellular buffer into the isolated mouse pancreas through a fine-gauge needle (30G x ½”) and a 1 ml syringe. The aim is to inflate the pancreas with the collagenase solution and sometimes multiple injections are needed to achieve this. The pancreas, in the collagenase solution, is then transferred to a 1.5 ml eppendorff and incubated for 6 minutes at 370C. We do not use any shaking during this incubation time.
The whole pancreas is then triturated vigorously by passing it up and down through a 1ml pipette (plastic pipette tip such as for a 1ml Gilson P1000 pipette) where the tip of the pipette has been cut to make a diameter of approximately 3 mm. If the pancreas goes through the pipette tip easily the diameter is too big. The aim is to sheer large fragments of tissue off the main body of the pancreas. Fragments are periodically collected in a 15 ml tube and fresh Na-rich media added. The process should take no longer than 5 minutes and lead to an accumulation of about 200 ml of pancreatic material settled to the bottom of 10 ml of Na-rich buffer.
This is then centrifuged to pellet the material, the supernatant discarded and the pellet resuspended in 3 ml of Na-rich media. Gentle trituration through a new uncut 1 ml pipette (again a plastic pipette tip such as for a 1ml Gilson P1000 pipette) for around 3 minutes is used to further break down the tissue fragment size. This is then centrifuged again, resuspended in 3 ml of Na-rich buffer and triturated again and this is the final suspension of exocrine fragments.
2.2 Paraformaldehdye fixation and confocal microscopy
The suspensions of freshly isolated pancreatic fragments (100ml) are plated onto poly-L-lysine coated glass coverslips and left for 5 minutes to allow the fragments to stick to the glass. Lysine fixable dye (final concentration 2mg/ml) is added, and mixed with the cell suspension, 5 minutes prior to cell stimulation. In a typical experiment we stimulate by adding 600 nM of acetylcholine to the cell suspension droplet for 2 minutes and then add atropine 1 mg/ml to stop the stimulation. The cells are then fixed with freshly prepared paraformaldehyde (8% at 4°C) added at a 50:50 volume with the cell suspension, usually 100 ml. This fixative is left for 20 minutes and then is washed with PBS for three times over a 10 minute time period.
If required for immunohistochemistry the tissue fragments are permeabilized with 1% Triton-X in phosphate buffered solution (PBS) for 5 minutes. This is followed by standard techniques of blocking and incubation with primary and secondary antibodies.
There are no special requirements for confocal microscopy. We typically use a 60x 1.35 na fluorescence objective and take 12 bit images of 1024 x 1024 pixels. For quantitative analysis we take care to fulfill the requirements of digital imaging including using the relevant pixel size for optimal resolution and working to maximize the full bit depth without saturation.
Figure 1 shows an example of the use of lysine fixable FITC-dextran extracellular dye in a tissue fragment stimulated with acetylcholine and fixed with paraformaldehyde. After fixation and Triton-X permeabilization the F-actin cytoskeleton was stained with rhodamine-phalloidin.
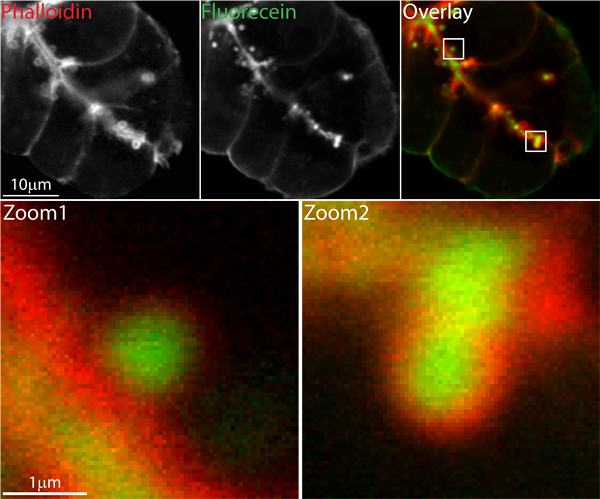
Figure 1. Confocal image of pancreatic fragment showing the characteristic F-actin coating that forms after zymogen granule fusion. Cells were bathed in lysine-fixable FITC dextran, then stimulated with acetylcholine. They were then paraformaldehyde fixed, Triton-X permeabilized and incubated in rhodamine-phalloidin to label F-actin. The upper low magnification images are 700x700 pixels with 50 nm pixel size.
Fluorescein (green) labels the lumen, which in this tissue fragment lies diagonally through the cells, and each individual fused zymogen granule. The F-actin (red) surrounds the lumen and, as shown in the zoomed in areas, surrounds each individual granule.
Figure 2 is another example image, taken from Bhat & Thorn (2011) (2), and shows pancreatic tissue fragments stimulated with acetylcholine and paraformaldehyde fixed. Here the extracellular dye was lysine fixable FITC dextran (green), F-actin was stained with TRITC-phalloidin and myosin 2A (lower) and myosin 2B (upper) were stained with Alexa-633 conjugated secondary antibodies (colour coded blue).
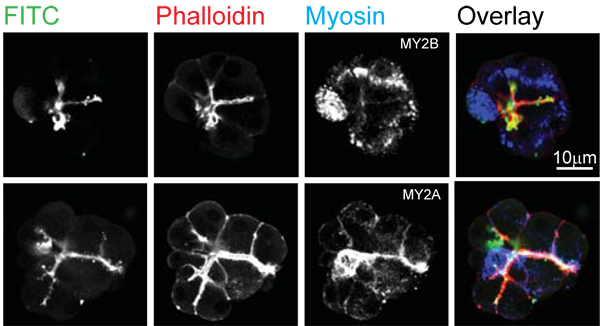
Figure 2. Confocal image of pancreatic fragment showing F-actin distribution and immunolocalization of myosin 2A and myosin 2B. Cells were bathed in lysine-fixable FITC dextran, then stimulated with acetylcholine. TRITC-phalloidin was used to label F-actin. The upper images show that myosin 2B is apparently not near the lumen. In contrast myosin 2A (lower images) is closely associated with the luminal F-actin cytoskeleton. Figure taken from Bhat & Thorn 2009 (2)
2.3 Live-cell two photon imaging
We typically place 100 ml of the freshly made suspension of pancreatic fragments onto a poly-l-lysine coated coverslip. After letting the fragments stick we then add (and mix) the extracellular fluorescent dyes. Routinely we use sulforhodamine B (8, 10) which is an inert dye that is intensely fluorescent with two-photon illumination. The intensity of the laser light and the gains on the detectors are adjusted to provide the best signal to noise characteristics. Usually we work with the photomultiplier gain at around 50% since this minimises noise. We then adjust the laser intensity (on the laboratory-built machine this is via adjustment of a pockel-cell) to maximise the signal but are careful to work within a range where cell damage doesn’t occur. Typical images, such as in the attached movie, show bright fluorescence surrounding the tissue fragment, bright fluorescence in the lumens and little to no fluorescence within the cell. When the cells are stimulated exocytosis of zymogen granules is observed as a sudden increase in fluorescence within each fused granule (see movie).
We capture images at frame rates of 30 frames per second. This resolves the fast rise times during dye entry into the granules. However, these fast rates are not necessary to track granule lifetimes and we routinely average frames together to give effective capture rates of 2 frames per second.
Image capture on our system is 3 channel, 8 bit, 400x600 pixels. With pixel sizes, dependent on experimental aims, adjusted from 6 pixels/mm to 15 pixels/mm. Analysis is performed off-line with Metamorph (Molecular Devices).
3. Notes
- Confocal microscopy of acinar lumens and fused granules requires that the extracellular lysine-fixable dyes cross link with the cell-surface proteins. This has led us to use paraformaldehyde at 8% with a 50:50 dilution with the cell suspension giving a 4% final concentration. Since the paraformaldehdye does not contain extracellular dye this method also dilutes the dye by 50%. Our experience is that any further dilution or washing with paraformaldehdye decreases the staining.
- We have used many different extracellular dyes in the live two photon experiments. One issue with dyes is that they bind to proteins and this affects the observed signal. In this way dyes not only enter fused granules but accumulate on the granule content that is within the granule. The kinetics of the fluorescence signal, at times that closely follow fusion of the granules, is therefore a combination of dye entry, dye accumulation and then loss of granule content (10).
- Deliberate choice of environmentally-sensitive extracellular dyes can be used to track ion changes in the lumen and the granules. For example, we have used the pH sensitive dye, HPTS, to show acid changes in the lumen associated with release of acid from zymogen granules (1).
4. References
- Behrendorff N, Floetenmeyer M, Schwiening C, and Thorn P. Protons Released During Pancreatic Acinar Cell Secretion Acidify the Lumen and Contribute to Pancreatitis in Mice. Gastroenterology 139: 1711-1715, 2010. PMID 20691184
- Bhat P and Thorn P. Myosin 2 Maintains an Open Exocytic Fusion Pore in Secretory Epithelial Cells. Molecular Biology of the Cell 20: 1795-1803, 2009. PMID 19158378
- Campos-Toimil M, Edwardson JM, and Thomas P. Real-time studies of zymogen granule exocytosis in intact rat pancreatic acinar cells. Journal of Physiology-London 528: 317-326, 2000. PMID 11034621
- Chen Y, Warner JD, Yule DI, and Giovannucci DR. Spatiotemporal analysis of exocytosis in mouse parotid acinar cells. American Journal of Physiology-Cell Physiology 289: C1209-C1219, 2005. PMID 16000640
- Gaisano HY. A hypothesis: SNARE-ing the mechanisms of regulated exocytosis and pathologic membrane fusions in the pancreatic acinar cell. Pancreas 20: 217-226, 2000. PMID 10766446
- Kasai H, Kishimoto T, Nemoto T, Hatakeyama H, Liu TT, and Takahashi N. Two-photon excitation imaging of exocytosis and endocytosis and determination of their spatial organization. Advanced Drug Delivery Reviews 58: 850-877, 2006. PMID 16996640
- Larina O, Bhat P, Pickett JA, Launikonis BS, Shah A, Kruger WA, Edwardson JM, and Thorn P. Dynamic regulation of the large exocytotic fusion pore in pancreatic acinar cells. Molecular Biology of the Cell 18: 3502-3511, 2007. PMID 17596517
- Nemoto T, Kimura R, Ito K, Tachikawa A, Miyashita Y, Iino M, and Kasai H. Sequential-replenishment mechanism of exocytosis in pancreatic acini. Nature Cell Biology 3: 253-258, 2001. PMID 11231574
- Segawa A and Riva A. Dynamics of salivary secretion studied by confocal laser and scanning electron microscopy. European Journal of Morphology 34: 215-219, 1996. PMID 8874099
- Thorn P and Parker I. Two phases of zymogen granule lifetime in mouse pancreas: ghost granules linger after exocytosis of contents. Journal of Physiology-London 563: 433-442, 2005. PMID 15637100
- Turvey MR and Thorn P. Lysine-fixable dye tracing of exocytosis shows F-actin coating is a step that follows granule fusion in pancreatic acinar cells. Pflugers Archiv-European Journal of Physiology 448: 552-555, 2004. PMID 15103465
- Williams JA. Regulation of acinar cell function in the pancreas. Curr Opin Gastroenterol 26: 478-483, 2010. PMID 20625287