
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2013.14
| Attachment | Size |
|---|---|
| 334.49 KB |
Introduction
In 1995 when Yoshida et al. coined the term “autoimmune pancreatitis” they listed several serologic and imaging features that helped them recognize the entity(10). These features formed the basis for the first diagnostic criteria proposed by the Japan Pancreas Society in 2002 (9). In these reports hypergammaglobulinemia and non-specific markers such as rheumatoid factor and antinuclear antibodies served as serologic markers of AIP. In 2001 Hamano et al. made the observation that elevated serum IgG4 levels were highly sensitive and specific for the diagnosis of autoimmune pancreatitis (AIP) (4). Even though this led to a rapid increase in the number of patients diagnosed with AIP, it soon became clear this was an inadequate biomarker when used in isolation to diagnose AIP. As the larger spectrum of disease became apparent the need for diagnostic criteria to distinguish AIP from other diseases including chronic pancreatitis, pancreatic cancer, and other systemic diseases was evident. Within one decade there were at least six versions of diagnostic criteria published by groups from Japan, Italy, the United States, and Korea (1-3, 5-7). Although the criteria were generally similar, there were major differences including the necessity of ERCP for diagnosis, and the inclusion or exclusion of criteria for other organ involvement and response to steroids. Unfortunately, these differences led to confusion for practicing clinicians and prevented comparison of results between studies.
International Consensus Diagnostic Criteria
In 2011 a multinational group met to develop diagnostic criteria that would be meaningful for both clinical and research purposes. They achieved a consensus that recognizes our current understanding of AIP, permits flexibility in diagnostic evaluation (e.g., reliance on histology vs. pancreatography), and acknowledges the two AIP subtypes. Importantly, although the typical clinical presentation of patients with AIP is obstructive jaundice, occasionally with a mass, the criteria also permit diagnosis in those with less common disease presentations and indeterminate imaging findings. These criteria are referred to as the International Consensus Diagnostic Criteria (ICDC) for AIP (8). The cardinal clinical features of AIP in the ICDC are based on pancreatic parenchymal imaging, pancreatic ductal imaging (i.e., ERP), serum IgG4 level, other organ involvement, histology of the pancreas, and response to steroid treatment.
Diagnostic components of International Consensus Diagnostic Criteria
Historically, pancreatic imaging findings (both parenchymal and ductal) were considered as the essential for diagnosis of AIP. Using ICDC, ERCP is not necessary for diagnosis, but rather parenchymal and ductal imaging are recognized as separate, yet complementary, criteria. The serum IgG4 level is appreciated as a more sensitive and specific disease marker than previously used serologies, which included total IgG, γ-globulin, and auto-antibody (ANA and RF) levels, and is therefore the preferred serologic test (Serlogic Abnormalities in Autoimmune Pancreatitis) . AIP is now recognized as the pancreatic manifestation of a multi-organ syndrome called IgG4-Related Disease (IgG4-RD). As a result, the presence of other organs commonly associated with IgG4-RD (e.g., biliary strictures located proximal to the intrapancreatic portion of the common bile duct, retroperitoneal fibrosis, and sialadenitis) are supportive findings for AIP, and referred to as other organ involvement (OOI) (Extra-pancreatic Features of Autoimmune Pancreatitis). Pancreatic histology obtained by core tissue biopsy (or a resected pancreatic specimen) is uniquely recognized as the “gold standard” for AIP diagnosis (Histology of Autoimmune Pancreatitis). This distinction is made on the basis that pathologists are able to accurately diagnose AIP independently of other clinical information (11). Finally, response to steroid treatment evidenced by resolution or marked improvement in radiographic features is recognized as an important criterion. Although not included in some of the initial diagnostic criteria schemes, it was later recognized that a significant portion of patients did not have the characteristic imaging findings, yet still responded to steroid therapy.
Use of International Consensus Diagnostic Criteria to diagnose autoimmune pancreatitis
ICDC are organized to allow the user to diagnose AIP along several potential pathways; i.e., characteristic imaging, characteristic histology, or response to steroid therapy. However, it all starts with review of pancreatic findings on cross-sectional imaging (CT or MRI). These are classified as typical of AIP (level 1) or atypical/indeterminate for AIP (Level 2). Next, available data supporting the diagnosis of AIP (i.e., collateral evidence) is considered in combination with the imaging findings. Collateral evidence (i.e., pancreatic ductal imaging, serum IgG4, other organ involvement, and response to treatment) is assigned one of two levels based on the strength of association with AIP (Table 1). To establish a definitive diagnosis of type 1 AIP varying strengths of collateral evidence are needed depending on imaging findings (Table 2). For example, in patients with typical parenchymal imaging for AIP (which is a relatively specific finding) any level of non-pancreatic collateral evidence secures a definitive diagnosis. Conversely, stronger collateral evidence is required when imaging is indeterminate for AIP. Using these criteria several combinations can be used to establish an AIP diagnosis, even without the need for histology or ERP.
One advantage of ICDC is the recognition of the two AIP subtypes. Type 2 AIP is generally characterized by the lack of serum IgG4 elevation and OOI, and is occasionally associated with inflammatory bowel disease. However, this profile is also present in some patients with type 1 AIP, so histology is the only means of establishing a definitive diagnosis of type 2 AIP. While the ICDC are specific for AIP, a subset of subjects with unequivocal steroid responsive pancreatic mass/enlargement who have no or minimal collateral evidence fail to meet diagnostic criteria for a definitive AIP subtype. Such patients are classified as AIP-not otherwise specified (AIP-NOS). When inflammatory bowel disease is present these patients are felt to likely have type 2 AIP and are classified as probable type 2 AIP.


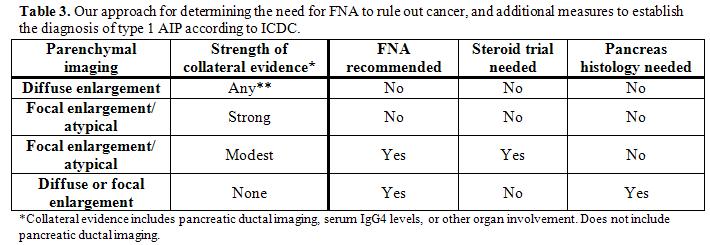
A practical approach to using the International Consensus Diagnostic Criteria
The complexity of the criteria used in the ICDC is necessary due to the protean disease presentations. Although it may initially appear too cumbersome for clinical use, with thoughtful consideration a practical approach for using the ICDC is possible. Since AIP is extremely rare, the responsibility of the clinician is primarily to exclude an alternative etiology (namely malignancy) rather than to establish an AIP diagnosis. Therefore, in most cases unless noninvasive studies (i.e., imaging and serum IgG4 levels) are characteristic for AIP, some form of cytology (obtained with or without core biopsy for histology) is necessary. In the absence of convincing evidence for malignancy additional testing can be pursued for cases in which AIP is suspected.
Our current approach for diagnosing AIP and distinguishing it from pancreatic cancer using ICDC is shown in Table 3. The initial step is to determine the likelihood of AIP based on pancreatic parenchymal imaging. When typical imaging (e.g., diffuse pancreatic enlargement with delayed enhancement of the parenchyma, with or without presence of a capsule sign) is present any non-ductal imaging collateral evidence (i.e. elevated serum IgG4 OR presence of OOI) will establish an AIP diagnosis. In these patients a diagnostic steroid trial and core biopsy of pancreas are unnecessary to support the diagnosis (although steroids are generally initiated for therapeutic purposes). On the other hand, if the pancreatic imaging shows focal/segmental enlargement or has atypical features (e.g., low-density mass, pancreatic duct dilation, or distal atrophy) the subsequent evaluation is dictated by the amount and strength of the collateral evidence. If there is strong collateral evidence (need 2 of the following: i.) pancreatic duct stricture without upstream dilation, ii.) serum IgG4 >2x upper limit of normal value, iii.) histologic demonstration of OOI or radiographic presence of proximal biliary disease or retroperitoneal fibrosis), then the diagnosis of AIP can be confirmed without additional measures. Conversely, if the collateral evidence is only modest, FNA is first recommended to rule out cancer, then a steroid treatment trial is needed to secure the AIP diagnosis. Modest collateral evidence would be satisfied with the presence of one of the following: i.) typical long or multiple pancreatic ductal strictures on ERP AND either serum IgG4 elevation or presence of OOI, ii.) serum IgG4 >2x upper limit of normal value, or iii.) histologic documentation of OOI or radiographic presence of proximal biliary disease or retroperitoneal fibrosis. It should be highlighted that diagnostic steroid trials are rarely needed, and are not recommended unless cancer has been excluded by FNA of a mass lesion. Repeat imaging is recommended after 2 weeks of steroid treatment, and if there is not significant improvement alternative etiologies should be considered. Finally, regardless of the nature of pancreatic imaging features, if there is no supportive collateral evidence for AIP, an FNA is recommended to exclude cancer and a core biopsy of the pancreas is needed to reach an AIP diagnosis.
Summary
AIP is an increasingly recognized clinical entity. Although an elevated serum IgG4 level is an important diagnostic clue, this is inadequate to establish a diagnosis independently and other collateral evidence is needed. The ICDC are recently published diagnostic criteria that help both the clinician and researcher accurately identify those with AIP using one of several combinations of key diagnostic features (pancreatic parenchymal imaging, pancreatic ductal imaging (i.e. ERP), serum IgG4 level, other organ involvement, histology of the pancreas, and response to steroid treatment). Since AIP is rare the clinician’s diagnostic approach must primarily focus on the exclusion of malignancy, and then on solidifying the AIP diagnosis.
References
- Chari ST, Smyrk TC, Levy MJ, Topazian MD, Takahashi N, Zhang L, Clain JE, Pearson RK, Petersen BT, Vege SS, and Farnell MB. Diagnosis of autoimmune pancreatitis: the Mayo Clinic experience. Clin Gastroenterol Hepatol 4: 1010-1016; quiz 1934, 2006. PMID: 16843735
- Chari ST, Takahashi N, Levy MJ, Smyrk TC, Clain JE, Pearson RK, Petersen BT, Topazian MA, and Vege SS. A diagnostic strategy to distinguish autoimmune pancreatitis from pancreatic cancer. Clin Gastroenterol Hepatol 7: 1097-1103, 2009. PMID: 19410017
- Frulloni L, Scattolini C, Falconi M, Zamboni G, Capelli P, Manfredi R, Graziani R, D'Onofrio M, Katsotourchi AM, Amodio A, Benini L, and Vantini I. Autoimmune pancreatitis: differences between the focal and diffuse forms in 87 patients. Am J Gastroenterol 104: 2288-2294, 2009. PMID: 19568232
- Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, Fukushima M, Nikaido T, Nakayama K, Usuda N, and Kiyosawa K. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 344: 732-738, 2001. PMID: 11236777
- Kim KP, Kim MH, Kim JC, Lee SS, Seo DW, and Lee SK. Diagnostic criteria for autoimmune chronic pancreatitis revisited. World J Gastroenterol 12: 2487-2496, 2006. PMID: 16688792
- Otsuki M, Chung JB, Okazaki K, Kim MH, Kamisawa T, Kawa S, Park SW, Shimosegawa T, Lee K, Ito T, Nishimori I, Notohara K, Naruse S, Ko SB, and Kihara Y. Asian diagnostic criteria for autoimmune pancreatitis: consensus of the Japan-Korea Symposium on Autoimmune Pancreatitis. J Gastroenterol 43: 403-408, 2008. PMID: 18600383
- Pearson RK, Longnecker DS, Chari ST, Smyrk TC, Okazaki K, Frulloni L, and Cavallini G. Controversies in clinical pancreatology: autoimmune pancreatitis: does it exist? Pancreas 27: 1-13, 2003. PMID: 12826899
- Shimosegawa T, Chari ST, Frulloni L, Kamisawa T, Kawa S, Mino-Kenudson M, Kim MH, Kloppel G, Lerch MM, Lohr M, Notohara K, Okazaki K, Schneider A, and Zhang L. International consensus diagnostic criteria for autoimmune pancreatitis: guidelines of the International Association of Pancreatology. Pancreas 40: 352-358, 2011. PMID: 21412117
- Society MotCCfAPotJP. Diagnostic criteria for autoimmune pancreatitis by the Japan Pancreas Society (2002). J Jpn Pancreas Soc 17: 585-587, 2002.
- Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, and Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci 40: 1561-1568, 1995. PMID: 7628283
- Zhang L, Chari S, Smyrk TC, Deshpande V, Kloppel G, Kojima M, Liu X, Longnecker DS, Mino-Kenudson M, Notohara K, Rodriguez-Justo M, Srivastava A, Zamboni G, and Zen Y. Autoimmune pancreatitis (AIP) type 1 and type 2: an international consensus study on histopathologic diagnostic criteria. Pancreas 40: 1172-1179, 2011. PMID: 21975436