
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2013.19
| Attachment | Size |
|---|---|
| 987.4 KB |
Introduction
IgG4-related disease (IgG4-RD) was identified and recognized as a multi-organ disease during the first decade of this century. The disease is characterized by histopathology and immunohistochemical staining patterns that are consistent across many organ systems (54). It is easier to indicate which organs are affected seldom (if ever) than to list all of the systems involved by this condition. To date, it has been rare to find confirmed cases of IgG4-RD affecting the brain parenchyma, muscle tissue, synovium, and bone marrow. Virtually every other organ is now known to be affected by IgG4-RD, with some organs affected more commonly than others.
IgG4-RD was first identified in the pancreas. During the 1990s, the concept of “autoimmune pancreatitis” (AIP) began to emerge from a variety of other names for a condition associated with sclerosing inflammation in that organ (68). “Sclerosing pancreatitis” was linked to elevated serum concentrations of IgG4 in 2001 (17). IgG4-RD was recognized as a systemic condition in 2003, when a variety of extra-pancreatic lesions were observed to occur in patients with AIP (20, 22).
IgG4-RD is now known to affect the pancreas, biliary tree, salivary glands, periorbital tissues (e.g., the lacrimal gland and retro-orbital space), kidneys, lungs, lymph nodes, meninges, aorta, breast, prostate, thyroid gland, pericardium, and skin (Table 1) (16, 18, 15, 59, 21, 11, 50, 65, 10, 72, 46, 62, 26, 23, 52, 56, 19, 71). The general pathology findings in any organ include a lymphoplasmacytic infiltrate, fibrosis that typically has a storiform pattern, obliterative phlebitis, modest tissue eosinophilia, and the tendency to form tumefactive lesions (14). This chapter is devoted to a review of the extra-pancreatic features of IgG4-RD that can be seen in those with type 1 AIP.
Nomenclature
The Organizing Committee of the 2011 Boston International Symposium on IgG4-RD recommended names for the individual organ system manifestations of this disease (50). These are shown in Table 2. The nomenclature system reinforces the concept that the same fundamental pathophysiologic processes are operative across organ systems, regardless of whether the role of IgG4 is primary or secondary. Individual organ involvement is referred to in a style that employs “IgG4-related-” as a prefix. As examples, the most common form of kidney involvement in IgG4-RD is termed IgG4-related tubulointerstitial nephritis, and eye manifestations of this condition are regarded collectively as IgG4-related ophthalmic disease.

Systemic Features
IgG4-RD typically has an indolent presentation. Features of the disease generally become manifest after months or even years. In addition, constitutional symptoms are subtle or absent in the majority of patients, who often feel relatively well even in the setting of multi-organ disease. Others, however, have anorexia and weight loss that becomes substantial over time. A minority of patients have more explosive presentations characterized by constitutional symptoms, fevers, and acute phase reactant elevations.
Tumefactive Lesions (Pseudotumors)
Patients with IgG4-RD are often suspected or even misdiagnosed initially as having a malignancy, because of the disease’s predilection for causing mass-forming lesions within organs. Many patients are subjected to pancreatic resection out of concern for pancreatic cancer. In addition, pseudotumors are reported commonly in the orbital region, salivary glands, lung, kidney, lymph nodes, retroperitoneum, and other organs (72, 71, 27). Many have an indolent course, but local tissue destruction including the erosion of bone and aortic aneurysms or dissections have been reported (52, 28, 37, 48, 5, 53)

Diffuse infiltrative lesions also occur in organs such as the meninges, skin, or aorta.
Inflammatory pseudotumors have been described in IgG4-RD involving the lung and central nervous system (10, 72, 71, 32), as well as in the orbit (29), salivary gland (66), paraspinal regions (6), and other tissue and organs. Because of the tendency of IgG4-RD to cause tumefactive lesions, immunostaining should be performed on all pseudotumors with significant infiltrates of plasma cells and fibrosis (61).
Multifocal Fibrosclerosis
A condition known as multifocal fibrosclerosis was identified first in the 1960s (9). Multifocal fibrosclerosis, often characterized by the simultaneous occurrence of other fibrotic syndromes such as Riedel’s thyroiditis, hypertrophic pachymeningitis (3, 31), retroperitoneal fibrosis, fibrosing mediastinitis (59), sclerosing mesenteritis (4), and orbital pseudotumor, is probably explained in most cases by IgG4-RD.
Allergic Disease
Allergic or atopic manifestations occur in approximately 50% of patients with IgG4-RD. Patients with IgG4-RD often have longstanding histories of allergic rhinitis, sinusitis, asthma, and other clinical features of this nature. Many patients have substantial elevations of serum IgE concentrations or peripheral eosinophilias that sometimes approach 25% of the total white blood cell count. Mild to moderate eosinophil infiltration is also typical of tissue lesions (14).
Eosinophilic angiocentric fibrosis
One subset of IgG4-RD associated with striking allergic features is eosinophilic angiocentric fibrosis (EAF), an uncommon tumefactive lesion of the orbit and the upper respiratory tract (41, 12). The histopathology of this condition is characterized by an overabundance of eosinophils as well as an admixture of lymphocytes and plasma cells. Small-caliber arterioles in EAF demonstrate “onion-skinning” – concentric layers of fibrosis – of the blood vessels. Patients with EAF share the broader tendency of IgG4-RD to form tumefactive lesions within involved organs.
Lymphadenopathy
Lymphadenopathy in IgG4-RD generally takes two forms. First, generalized lymphadenopathy can be the major or sole component of the clinical presentation. Patients with IgG4-related lymphadenopathy of this nature are often constitutionally well, at least for prolonged periods. Second, involvement of lymph nodes as localized disease adjacent to a specific organ affected by IgG4-RD (e.g., the pancreas) is common.
Patients with IgG4-related lymphadenopathy often undergo serial lymph node biopsies to exclude lymphoma, sarcoidosis, multicentric Castleman’s disease, or disseminated malignancies. Rendering the diagnosis of IgG4-RD purely on the basis of lymph node pathology is difficult at the present time, however, because the histology of lymph nodes in this setting is remarkably variable (47). Clinicians must seek evidence of disease in other organ systems typically involved by IgG4-RD in order to be confident of the diagnosis. Increased numbers of IgG4+ plasma cells, of course, are universal, but the storiform fibrosis so common in other types of IgG4-RD organ involvement is unusual in IgG4-related lymphadenopathy. Lymph node biopsies in most cases are reported as “reactive follicular hyperplasia,” and specific stains for IgG4 generally not performed because the diagnosis is not considered.
Specific Organ Involvement by Body Region
The discussion of specific organ involvement is divided by the different major body regions: the head and neck, the chest, the abdomen/retroperitoneum, and miscellaneous.
Head & Neck
Meninges
A study of fifteen cases of idiopathic hypertrophic pachymeningitis found that IgG4-RD, linked to four of the cases (27%), was the most common disease association (62). Other causes in that series were granulomatosis with polyangiitis (3 cases) and miscellaneous other diagnoses including sarcoidosis, lymphoma, giant cell arteritis, and rheumatoid arthritis. This study confirmed an earlier one that had demonstrated IgG4-RD to be the cause of five out of ten cases of idiopathic lymphoplasmacytic meningeal inflammation (31).
Pituitary gland
The presence of IgG4-RD in other organs and elevated serum IgG4 concentrations are common features of IgG4-related hypophysitis and often constitute a major clue to the diagnosis of IgG4-related hypophysitis. The radiologic findings in this entity include a pituitary mass or thickened pituitary stalk. Most IgG4-RD patients with hypophysitis are middle-aged men who present with various degrees of anterior or posterior hypopituitarism. IgG4-RD must be differentiated from sarcoidosis, granulomatosis with polyangiitis (formerly Wegener’s), histiocytosis, and lymphoma as causes of hypophysitis. Both glucocorticoid therapy and rituximab can lead to resolution of imaging abnormalities and clinical signs of pituitary insufficiency.
Salivary Glands
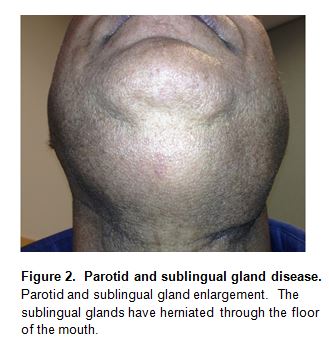
“Küttner’s tumor”, another disorder described originally in the 1890s (30), consists of a tumorous swelling of the submandibular glands (Figure 1). Bilateral submandibular gland swelling in the absence of stones within Wharton’s duct is commonly associated with IgG4-RD (16, 8). In contrast to Sjögren’s syndrome (SjS), in which the degree of parotid enlargement is often dramatically out of proportion to that of the submandibular glands, the opposite pattern is more typical of IgG4-related sialadenitis. Submandibular gland disease often occurs in IgG4-RD in the absence of any clinical evidence of parotid gland enlargement, but parotid disease in IgG4-RD is also described (Figure 2). Salivary gland involvement consists of firm, nodular swelling that is generally symmetrical and associated with pain, tenderness, and decreased saliva production. The sublingual glands can also be affected by IgG4-RD (Figure 2).
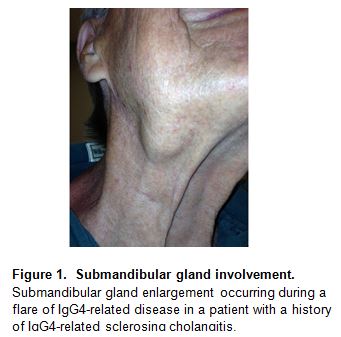
The triad of swelling in the submandibular, parotid, and lacrimal glands, first described in 1892, was termed “Mikulicz disease” for more than 100 years (34, 35). For decades, Mikulicz disease was believed to be a variant of SjS (35, 36), and even today patients with IgG4-RD are continually misdiagnosed with SjS. IgG4-RD can be differentiated clearly from SjS on the basis of clinical, serological, and pathological findings (67).
Minor salivary glands of the lip may be affected, as demonstrated by lip biopsy (2). Minor salivary gland disease can be present even when the glands are macroscopically normal. The sensitivity and specificity of minor salivary gland biopsy in patients with IgG4-RD have not been well defined.
Ophthalmic disease
The orbital tissues are perhaps the most commonly affected region of the body in IgG4-RD. IgG4-RD accounts for a substantial percentage – at least 25% – of the cases of “idiopathic” orbital inflammation, a differential diagnosis that includes lymphoma, granulomatosis with polyangiitis, Graves’ orbitopathy, and other conditions (56).
The ocular structures affected by IgG4-RD include the lacrimal glands (dacryoadenitis) (Figure 3), the nasolacrimal duct, and the retro-bulbar region that is frequently involved by orbital pseudotumor (Figure 4), and the extra-ocular muscles (a condition often termed “orbital myositis”) (64, 37, 46, 7). As noted, many cases of IgG4-related dacryoadenitis are accompanied by salivary gland disease. In some patients with IgG4-related ophthalmic disease, the process extends beyond the orbit and tracks along the course of the trigeminal nerve (37, 24).
IgG4-RD accounts for a substantial percentage – at least 25% – of the cases of “idiopathic” orbital inflammation, a differential diagnosis that includes lymphoma, granulomatosis with polyangiitis, Graves’ orbitopathy, and other conditions (56).
The ocular structures affected by IgG4-RD include the lacrimal glands (dacryoadenitis) (Figure 3), the nasolacrimal duct, and the retro-bulbar region that is frequently involved by orbital pseudotumor (Figure 4), and the extra-ocular muscles (a condition often termed “orbital myositis”) (64, 37, 46, 7). As noted, many cases of IgG4-related dacryoadenitis are accompanied by salivary gland disease. In some patients with IgG4-related ophthalmic disease, the process extends beyond the orbit and tracks along the course of the trigeminal nerve (37, 24).
Thyroid gland
Riedel’s thyroiditis has been identified as a manifestation of IgG4-RD (11). Although thyroid gland biopsies from patients with advanced disease simply demonstrate glandular fibrosis, biopsies from early in the disease course demonstrate the classic pathologic hallmarks of IgG4-RD. The fibrosing variant of Hashimoto’s thyroiditis also appears to be part of the IgG4-RD spectrum, at least in a percentage of cases (13). Finally, it has been postulated that classic Hashimoto’s thyroiditis, distinct from the fibrosing variant discussed above, also falls within the IgG4-RD spectrum and might account for the high frequency of hypothyroidism in AIP. This putative “IgG4-related thyroiditis” is said to have a lower likelihood of anti-thyroid autoantibodies compared with Hashimoto’s thyroiditis, a low likelihood of diffuse goiter, and a favorable response to glucocorticoid treatment. However, relatively few such cases have been examined histopathologically and this hypothesis remains controversial and unconfirmed.

Chest
Lung
Pulmonary involvement in IgG4-RD is protean in its scope, and understanding of the nature and extent of lung and airway disease continues to evolve. At least six major patterns of IgG4-related pulmonary disease are described on the basis of radiological and histological findings (72, 70, 58, 49): 1) nodules; 2) thickening of the bronchovascular bundle (Figure 5); 3) alveolar interstitial disease (with honeycombing, bronchiectasis, and diffuse ground-glass opacities); 4) rounded ground-glass opacities; 5) pleural lesions associated with severely thickenedvisceral or parietal pleura with diffuse sclerosing inflammation; and, 6) airway lesions leading to narrowing.
Some patients with IgG4-related lung disease experience cough, hemoptysis, dyspnea, pleural effusion, or chest discomfort (15, 56, 58). In others, however, the presence of lung disease is asymptomatic and discovered only incidentally upon imaging.
Thoracic Aorta
IgG4-RD appears to account for at least 10% of cases of inflammatory aortitis involving the
thoracic aorta (51). The lymphoplasmacytic aortitis associated with IgG4-RD must be distinguished from the granulomatous inflammation that accompanies a somewhat larger subset of cases diagnosed as giant cell aortitis. The entity referred to by the descriptive pathologic term “chronic sclerosing aortitis” probably represents IgG4-RD in the great majority of cases. The term “isolated aortitis” should generally be regarded as unsatisfactory now without thorough attempts at differentiating IgG4-related aortitis from giant cell aortitis and other causes of aortic inflammation.
Retroperitoneum & Abdomen
Manifestations of IgG4-RD within many of the abdominal organs are addressed in detail in other sections of this publication; namely, those associated with pancreatic, biliary, liver, or gallbladder disease (insert links to Chapter 5. CT and MR features of autoimmune pancreatitis, Chapter 6 ERCP features of autoimmune pancreatitis). This section, therefore, focuses on retroperitoneal organs.
Chronic Periaortitis
Chronic periaortitis is now regarded as the umbrella term for inflammation in the retroperitoneum and peri-aortic regions that includes such entities as retroperitoneal fibrosis, inflammatory abdominal aortic aneurysm, and perianeurysmal fibrosis (39). IgG4-RD is now known to cause well over half of the cases previously regarded as “idiopathic” retroperitoneal fibrosis (Figure 6) (25, 73). When detected early, biopsies from patients with retroperitoneal fibrosis whose disease demonstrate histopathological and immunohistochemical features that are diagnostic of IgG4-RD (73). In contrast, biopsies performed in patients at a later phase of disease simply show fibrosis, because they represent a more advanced IgG4-RD stage in which the fibrotic features have become predominant. Nevertheless, the storiform morphology of the fibrosis can still be highly suggestive of IgG4-RD. In such cases, the IgG4/total IgG ratio within tissue rather than the number of IgG4-positive plasma cells/high-power field is often a key to recognizing IgG4-related retroperitoneal fibrosis (25).
The first study to connect retroperitoneal fibrosis and the entity now termed IgG4-RD was that of Hamano et al. (18), who reported three patients with retroperitoneal fibrosis and elevated serum IgG4 concentrations. Zen et al. reported that 10 of 17 retroperitoneal fibrosis patients had both elevated serum IgG4 concentrations and histopathological features typical of IgG4-RD (73).
Moreover, IgG4-related retroperitoneal fibrosis had an overwhelming tendency to occur in males. This tendency was confirmed by Khosroshahi et al. (25), who identified the histopathological and immunohistochemistry signature of IgG4-RD in the biopsies of 13 of 23 cases of “idiopathic” retroperitoneal fibrosis.
Inflammatory abdominal aortic aneurysm is also associated with IgG4-RD (23,53). Patients with inflammatory abdominal aortic aneurysms have clinical, demographic, and radiologic features that are distinct from those of patients with classic atherosclerotic aortic aneurysms.


Kidney
The radiologic appearance of the kidneys in IgG4-related renal disease can take several forms. Diffuse enlargement accompanied by parenchymal hypodensities may occur. Focal tumefactive lesions may also develop, mimicking renal cell carcinoma (10). More than one such lesion within any kidney can occur. A thickening of the renal pelvis has also been described (40). Finally, following a period of IgG4-related renal disease, pronounced atrophy of the kidneys can ensue. This atrophy can occur even in patients whose disease has appeared to respond to therapy with glucocorticoids (42).
The most common histopathological correlate of these radiologic findings is tubulointerstitial nephritis (TIN), which consists of patchy or diffuse tubulointerstitial lymphoplasmacytic infiltrates within a fibrotic interstitium (44). The clinical manifestations of IgG4-related TIN include proteinuria, hematuria, and decreased kidney function, sometimes culminating in end-stage renal disease (58, 66).
Patients with IgG4-related renal disease are typically hypocomplementemic and often profoundly so (43). The basis for this phenomenon remains incompletely defined, because the IgG4 molecule itself does not activate complement avidly. Nevertheless, renal biopsies in IgG4-related TIN demonstrate immune complexes that consist in part of IgG4 (10). The TIN associated with IgG4-RD can be differentiated histopathologically and immunohistochemically from other causes of TIN (40, 38). The great majority of patients with IgG4-related renal disease also have disease that is manifest in other organs, as well (42).
A second form of renal disease – membranous glomerulonephritis (MGN) – has also been described in IgG4-RD, although this lesion is decidedly less common than TIN (42, 1). Nephrotic-range proteinuria can occur in IgG4-related MGN. IgG4-related MGN comprises a disorder that is distinct from idiopathic MGN, even though the autoantibody associated with the latter disorder is also principally of the IgG4 subclass. The MGN associated with IgG4-RD is probably secondary to immune complex deposition rather than to the usual destructive inflammatory process that characterizes other organ involvement by this condition.
Miscellaneous Organ Involvement
Miscellaneous organs in which the typical histopathological features of IgG4-RD have been reported include the skin (26), prostate gland (1, 70, 61), and pericardium (55). In all of these reports, the patients’ clinical features have been associated with disease in more classic IgG4-RD organs, as well. The true frequency with which IgG4-RD affects these organs is not known for certain, because the presence of disease there can be extremely subtle. In the skin, for example, IgG4-RD causes flesh-colored or erythematous papules that are often asymptomatic. Similarly, symptoms of prostatism in middle-aged to elderly males are likely to be diagnosed as benign prostatic hypertrophy. IgG4-related prostate disease, however, is known to cause symptoms of “benign prostatic hypertrophy” in men in their 30s.
Summary
Type 1 AIP is the pancreatic manifestation of a systemic disease referred to as IgG4-RD. The current diagnostic criteria for AIP recognize typical other organ involvement including proximal biliary strictures, retroperitoneal fibrosis, symmetric enlargement of salivary/lacrimal glands, and renal disease. However, manifestations and currently is recognized to involve essentially any organ. Histology is important not only to identify diagnostic features of IgG4-RD, but also to exclude malignancy in those with mass-forming lesions.
References
- Alexander MP, Larsen CP, Gibson IW et al. Membranous glomerulonephritis in IgG4-related disease. Kidney Int 83(3):455-62,2013. PMID: 23254897
- Baer AN, Gourin CG, Westra WH, Cox DP, Greenspan JS, Daniels TE. Rare diagnosis of IgG4-related systemic disease by lip biopsy in an international Sjogren syndrome registry. Oral Surg Oral Med Oral Pathol Oral Radiol 115(3):e34-9; 2013. PMID: 23146570
- Chan SK, Cheuk W, Chan KT, Chan JK. IgG4-related sclerosing pachymeningitis: a previously unrecognized form of central nervous system involvement in IgG4-related sclerosing disease. Am J Surg Pathol 33(8):1249-52, 2009. PMID: 19561447
- Chen TS, Montgomery EA. Are tumefactive lesions classified as sclerosing mesenteritis a subset of IgG4-related sclerosing disorders? J Clin Pathol 61:1093-7, 2008. PMID: 18682417
- Cheuk W, Chan JK. IgG4-related sclerosing disease: a critical appraisal of an evolving clinicopathologic entity. Adv Anat Pathol 17(5):303-32; 2010. PMID: 20733352
- Cheuk W, Tam FKY, Chan ANH et al. Idiopathic cervical fibrosis: A new member of IgG4-related sclerosing diseases. Am J Surg Pathol 34(11): 1678-85, 2010. PMID: 20871392
- Cheuk W, Yuen HK, Chan JK. Chronic sclerosing dacryoadenitis: part of the spectrum of IgG4-related sclerosing disease? Am J Surg Pathol 31(4):643-5, 2007. PMID: 17414116
- Chow TL, Chan TT, Choi CY, Lam SH. Kuttner's tumour (chronic sclerosing sialadenitis) of the submandibular gland: a clinical perspective. Hong Kong Med J 14(1):46-9, 2008. PMID: 18239243
- Comings DE, Skubi KB, Van Eyes J, Motulsky AG. Familial multifocal fibrosclerosis. Findings suggesting that retroperitoneal fibrosis, mediastinal fibrosis, sclerosing cholangitis, Riedel's thyroiditis, and pseudotumor of the orbit may be different manifestations of a single disease. Ann Intern Med 66:884-92, 1967. PMID: 6025229
- Cornell LD, Chicano SL, Deshpande V, Collins AB, Selig MK, Lauwers GY et al. Pseudotumors due to IgG4 immune-complex tubulointerstitial nephritis associated with autoimmune pancreatocentric disease. Am J Surg Pathol 31:1586-97, 2007. PMID: 17895762
- Dahlgren M, Khosroshahi A, Nielsen GP, Deshpande V, Stone JH. Riedel's thyroiditis and multifocal fibrosclerosis are part of the IgG4-related systemic disease spectrum. Arthritis Care Res (Hoboken) 62:1312-8; 2010. PMID: 20506114
- Deshpande V, Khosroshahi A, Nielsen GP, Hamilos DL, Stone JH. Eosinophilic Angiocentric Fibrosis Is a Form of IgG4-related Systemic Disease. Am J Surg Pathol 35(5):701-6, 2011. PMID: 21502911
- Deshpande V, Khosroshahi A, Nielsen GP, Stone JH. Fibrosing variant of Hashimoto’s thyroiditis is a IgG4-related disease. J Clin Pathol 65(8):725-8, 2012. PMID: 22659333
- Deshpande V, Zen Y, Ferry JA et al. Consensus Statement on the Pathology of IgG4-Related Disease. Mod Pathol 25(9):1181-92, 2012. PMID: 22596100
- Fukukura Y, Fujiyoshi F, Nakamura F, Hamada H, Nakajo M. Autoimmune pancreatitis associated with idiopathic retroperitoneal fibrosis. AJR Am J Roentgenol 181(4):993-5; 2003. PMID: 16936451
- Geyer JT, Ferry JA, Harris NL, Stone JH, Zukerberg LR, Lauwers GY, et al. Chronic sclerosing sialadenitis (Küttner tumor) is an IgG4-associated disease. Am J Surg Pathol 34:202-10, 2010. PMID: 20061932
- Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 344(10):732-8, 2001. PMID: 11450670
- Hamano H, Kawa S, Ochi Y, Unno H, Shiba N, Wajiki M, et al. Hydronephrosis associated with retroperitoneal fibrosis and sclerosing pancreatitis. Lancet 359(9315):1403-4, 2002. PMID: 11978339
- Hamed G, Tsushima K, Yasuo M, Kubo K, Yamazaki S, Kawa S, et al. Inflammatory lesions of the lung, submandibular gland, bile duct and prostate in a patient with IgG4-associated multifocal systemic fibrosclerosis. Respirology 12(3):455-7, 2007. PMID: 17539856
- Kamisawa T, Funata N, Hayashi Y, Eishi Y, Koike M, Tsuruta K, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol 38:982-4, 2003. PMID: 14614606
- Kamisawa T, Okamoto A, Funata N. Clinicopathological features of autoimmune pancreatitis in relation to elevation of serum IgG4. Pancreas 31:28-31; 2005. PMID: 15968244
- Kamisawa T, Okamoto A. Autoimmune pancreatitis: proposal of IgG4-related sclerosing disease. J Gastroenterol 41:613-25, 2006. PMID: 16932997
- Kasashima S, Zen Y, Kawashima A, Endo M, Matsumoto Y, Kasashima F. A new clinicopathological entity of IgG4-related inflammatory abdominal aortic aneurysm. J Vasc Surg 49:1264-71, 2009. PMID: 19217746
- Katsura M, Morita A, Horiuchi H, Ohtomo K, Machida T. IgG4-Related Inflammatory Pseudotumor of the Trigeminal Nerve: Another Component of IgG4-Related Sclerosing Disease? AJNR Am J Neuroradiol 32:E150-2, 2011. PMID: 20864523
- Khosroshahi A, Carruthers M, Stone JH, Shingare S, Sainani N, Deshpande V. Re-thinking Ormond’s Disease: A study of “idiopathic” and secondary cases of retroperitoneal fibrosis in the era of IgG4-related disease. Medicine (Baltimore) 92(2):82-91,2013. PMID: 23429355
- Khosroshahi A, Carruthers MD, Deshpande V, Leb L, Reed J, Stone JH. Cutaneous IgG4-related disease. American Journal of Medicine 124 (October): e7-8, 2011.
- Khosroshahi A, Stone JH. A clinical overview of IgG4-related systemic disease. Curr Opin Rheumatol 23(1):57-66, 2011. PMID: 21124086
- Khosroshahi A, Stone JR, Pratt DS, Deshpande V, Stone JH. Painless jaundice with serial multi-organ dysfunction. Lancet 25;373(9673):1494, 2009. PMID: 19394539
- Kishi K, Fujii T, Kohno T, Yoshimura K. Inflammatory pseudotumour affecting the lung and orbit. Respirology 14(3):449-51, 2009. PMID: 19353778
- Küttner H. Über entzündliche tumoren der submaaxillär-speicheldrüse. Beitr Klin Chir 15:815–834, 1896. PMID:
- Lindstrom KM, Cousar JB, Lopes MB. IgG4-related meningeal disease: clinico-pathological features and proposal for diagnostic criteria. Acta Neuropathol 120(6):765-76, 2010. PMID: 20844883
- Lui PC, Fan YS, Wong SS, Chan AN, Wong G, Chau TK, et al. Inflammatory pseudotumors of the central nervous system. Hum Pathol. 40(11):1611-7, 2009. PMID: 19656549
- Mikulicz J. Concerning a peculiar symmetrical disease of the lacrimal and
- Mikulicz J. Über eine eigenartige symmetrische erkränkung der Thränen- und Mundspeicheldrüsen. In: Billroth T, editor. Beiträge zur chirurgie festschrift gewidmet. Stuttgart: Ferdinand Enke. 610–630, 1892.
- Morgan WS, Castleman B. A clinicopathologic study of Mikulicz's disease. Am J Pathol 29(3):471-503, 1953. PMID: 13040489
- Morgan WS. The probable systemic nature of Mikulicz's disease and its relation to Sjögren's syndrome. N Engl J Med 251(1):5-10, 1954. PMID: 13176645
- Narain S, Wallace ZS, Deshpande V, Carruthers R, Liebsch N, Stone JH. IgG4-Related Systemic Disease (IgG4-RD): An Emerging Condition and Novel Cause Of Saddle-Nose Deformity. (Submitted).
- Nishi S, Imai N, Yoshida K, Ito Y, Saeki T. Clinicopathological findings of immunoglobulin G4-related kidney disease. Clin Exp Nephrol 15(6):810-9, 2011. PMID: 21870078
- Palmisano A, Vaglio A. Chronic periaortitis: a fibro-inflammatory disorder. Best Pract Res Clin Rheumatol 23(3):339-53, 2009. PMID: 19508942
- Raissian Y, Nasr SH, Larsen CP, Colvin RB, Smyrk TC, Takahashi N, et al. Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol 22(7):1343-52, 2011. PMID: 21719792
- Roberts PF, McCann BG. Eosinophilic angiocentric fibrosis of the upper respiratory tract: a mucosal variant of granuloma faciale? A report of three cases. Histopathology 9(11):1217-25, 1985. PMID: 4085985
- Saeki T, Kawano M, Mizushima I, Yamamota M, Wada Y, Nakashima H et al. The clinical course of patients with IgG4-related kidney disease. Kidney Int 2013; Epub ahead of print. PMID: 23698232
- Saeki T, Nishi S, Imai N, Ito T, Yamazaki H, Kawano M, et al. Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis. Kidney Int 78(10):1016-23, 2010. PMID: 23698232
- Saeki T, Nishi S, Ito T, Yamazaki H, Miyamura S, Emura I, et al. Renal lesions in IgG4-related systemic disease. Intern Med 46(17):1365-71, 2007. PMID: 17827834
- Sato Y, Ohshima K, Ichimura K, Sato M, Yamadori I, Tanaka T, et al. Ocular adnexal IgG4-related disease has uniform clinicopathology. Pathol Int 58(8):465-70, 2008. PMID: 18705764
- Sato Y, Yoshino T. IgG4-related lymphadenopathy. Int J Rheumatol 2012:572539, 2012. PMID: 22719769
- Schiffenbauer AI, Gahl WA, Jaffe E, Wahl C, Khosroshahi A, Deshpande V, Hoffman R, Stone JH, Gill F. IgG4-related disease masquerading as recurrent mastoiditis. The Laryngologist 122(3):681-4, 2012. PMID: 22252885
- Shrestha B, Sekiguchi H, Colby TV, Graziano P, Aubry MC, Smyrk TC, et al. Distinctive pulmonary histopathology with increased IgG4-positive plasma cells in patients with autoimmune pancreatitis: report of 6 and 12 cases with similar histopathology. Am J Surg Pathol 33(10):1450-62, 2009. PMID: 19623032
- Stone JH, Caruso PA, Deshpande V. Case records of the Massachusetts General Hospital. Case 24-2009. A 26-year-old woman with painful swelling of the neck. N Engl J Med 361:511-8, 2009. PMID: 19641208
- Stone JH, Khosroshahi A, Deshpande V, Chan JKC, Heathcote JG, Aalberse R et al. IgG4-Related Disease: Recommendations for the Nomenclature of this Condition and its Individual Organ System Manifestations. Arthritis & Rheumatism 64(10):3061-7, 2012. PMID: 22736240
- Stone JH, Khosroshahi A, Deshpande V, Stone JR. IgG4-related systemic disease accounts for a significant proportion of thoracic lymphoplasmacytic aortitis cases. Arthritis Care Res (Hoboken) 62(3):316-22, 2010. PMID: 20391477
- Stone JH, Khosroshahi A, Hilgenberg A, Spooner A, Isselbacher EM, Stone JR. IgG4-related systemic disease and lymphoplasmacytic aortitis. Arthritis Rheum 60:3139-45, 2009. PMID: 19790067
- Stone JH, Patel V, Oliveira G, Stone JR. A 60 year-old man with abdominal pain and multiple aortic aneurysms. N Engl J Med 367(24):2335-46, 2012. PMID: 23234517
- Stone JH, Zen Y, Deshpande, V. IgG4-related disease. N Engl J Med 366: 539-51, 2012. PMID: 22316447
- Sugimoto T, Morita Y, Isshiki K, et al. Constrictive pericarditis as an emerging manifestation of hyper-IgG4 disease. Int J Cardiol 130:e100–e101, 2008. PMID: 17727980
- Takahira M, Ozawa Y, Kawano M et al. Clinical Aspects of IgG4-Related Orbital Inflammation in a Case Series of Ocular Adnexal Lymphoproliferative Disorders. Int J Rheumatol 635473, 2012. PMID: 22548072
- Takeda S, Haratake J, Kasai T, Takaeda C, Takazakura E. IgG4-associated idiopathic tubulointerstitial nephritis complicating autoimmune pancreatitis. Nephrol Dial Transplant 19(2):474-6, 2004. PMID: 14736977
- Taniguchi T, Ko M, Seko S, Nishida O, Inoue F, Kobayashi H, et al. Interstitial pneumonia associated with autoimmune pancreatitis. Gut. 53(5):770, 2004. PMID: 15082601
- Taniguchi T, Kobayashi H, Fukui S, Ogura K, Saiga T, Okamoto M. A case of multifocal fibrosclerosis involving posterior mediastinal fibrosis, retroperitoneal fibrosis, and a left seminal vesicle with elevated serum IgG4. Hum Pathol 37:1237-9, 2006. PMID: 16938531
- Uehara T, Hamano H, Kawakami M, et al. Autoimmune pancreatitis-associated prostatitis: distinct clinicopathological entity. Pathol Int 58:118–125, 2008. PMID: 18199162
- Umehara H, Okazaki K, Masaki Y et al. A novel clinical entity, IgG4-related disease (IgG4-RD): General concepts and details. Mod Rheumatol 22(1):1-14, 2012. PMID: 21881964
- Wallace ZS, Carruthers MN, Khosroshahi A, Carruthers R, Shingare S, Despande V, and Stone JH. Hypertrophic Pachymeningitis: IgG4-related disease as a common etiology. Medicine (Baltimore) 92:206-16, 2013. PMID: 23793110
- Wallace ZS, Deshpande V, Stone JH. Ophthalmic manifestations of IgG4-related disease: Single-center experience and review of the literature. (Submitted).
- Wallace ZS, Khosroshahi A, Jakobiec FA, Deshpande V, Hatton MP, Ritter J, Ferry JA, Stone JH. IgG4-Related Systemic Disease as a Cause of "Idiopathic" Orbital Inflammation, Including Orbital Myositis and Trigeminal Nerve Involvement. Survey of Ophthalmology 57: 26-33, 2012. PMID: 22018678
- Watson SJ, Jenkins DA, Bellamy CO. Nephropathy in IgG4-related systemic disease. Am J Surg Pathol 30(11):1472-7, 2006. PMID: 17063091
- Yamamoto H, Yamaguchi H, Aishima S, Oda Y, Kohashi K, Oshiro Y et al. Inflammatory myofibroblastic tumor versus IgG4-related sclerosing disease and inflammatory pseudotumor: a comparative clinicopathologic study. Am J Surg Pathol 33(9):1330-40, 2009. PMID: 19718789
- Yamamoto M, Takahashi H, Ohara M, Suzuki C, Naishiro Y, Yamamoto H, et al. A new conceptualization for Mikulicz’s disease as an IgG4-related plasmacytic disease. Mod Rheumatol 16:335–40, 2006. PMID: 17164992
- Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci 40:1561-8, 1995. PMID: 7628283
- Yoshimura Y, Takeda S, Ieki Y, et al. IgG4-associated prostatitis complicating autoimmune pancreatitis. Intern Med 45:897–901, 2006. PMID: 16946571
- Zen Y, Inoue D, Kitao A, Onodera M, Abo H, Miyayama S, et al. IgG4-related lung and pleural disease: a clinicopathologic study of 21 cases. Am J Surg Pathol 33(12):1886-93, 2009. PMID: 19898222
- Zen Y, Kasahara Y, Horita K, Miyayama S, Miura S, Kitagawa S, et al. Inflammatory pseudotumor of the breast in a patient with a high serum IgG4 level: histologic similarity to sclerosing pancreatitis. Am J Surg Pathol 29:275-8, 2005. PMID: 15644785
- Zen Y, Kitagawa S, Minato H, Kurumaya H, Katayanagi K, Masuda S et al. IgG4-positive plasma cells in inflammatory pseudotumor (plasma cell granuloma) of the lung. Hum Pathol 36:710-7, 2005. PMID: 16084938
- Zen Y, Onodera M, Inoue D, Kitao A, Matsui O, Nohara T, et al. Retroperitoneal fibrosis: a clinicopathologic study with respect to immunoglobulin G4. Am J Surg Pathol 33(12):1833-9, 2009. PMID: 19950407