
Methods Type:
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2011.33
| Attachment | Size |
|---|---|
| 303.17 KB |
1. Introduction
The pancreas expresses and secretes various triglyceride lipases.(22) The predominant lipase is colipase-dependent pancreatic triglyceride lipase (PTL). PTL is the archetype of a lipase gene family that includes PTL homologues, pancreatic lipase-related proteins 1 and 2 (PLRP1 and PLRP2).(10) PLRP1 and PLRP2 are each present in pancreatic secretions. Also present is carboxyl ester lipase (CEL), alternatively known as bile-salt stimulated lipase (BSSL).(6) PTL strongly prefers triglyceride substrates over other potential substrates such as phospholipids or galactolipids. In contrast, PLRP2 and CEL have a broader substrate specificity that includes triglycerides. PLRP1 has no known lipase activity but it may modulate the activity of PTL.(3) Although each of these lipases can be isolated from pancreatic secretions or from the gland, most recent structure-function studies have utilized recombinant proteins purified from either baculovirus or Pichia pastoris expression systems. This chapter will describe methods for the production and purification of pancreatic lipases in those systems with emphasis on the P. pastoris system. Finally, the most common assay methods for triglyceride lipases will be described.
2. Expression
2.1 Baculovirus Expression Systems:
The first attempts to produce large quantities of pancreatic lipases for structure-function studies utilized baculovirus expression systems.(13, 18, 19) The system has several advantages for expression of recombinant proteins.(21) In combination with insect cells, baculovirus can produce high levels of recombinant lipases with appropriate post-translational modifications. Baculovirus have a limited host range, mainly insects, and are safe for vertebrates. The insect cells are relatively easy to culture and will grow in serum-free media. The original descriptions of pancreatic lipase production in the baculovirus system used the PVL1392 transfer vector, linearized genomic DNA from Autographa californica nuclear polyhedrosis virus (AcMNPV) and insect cells from Spodoptera frugoperda (Sf9). Since the original descriptions of lipase expression in baculovirus systems, multiple improvements have been made in baculovirus expression systems.(21) Transfer vectors have been improved as have methods for purification of recombinant virus. The introduction of bacterial artificial chromosomes containing the genome of the baculovirus AcMNPV, bacmids, allows the manipulation of the viral genome in bacteria. Kits utilizing modern technology are available (Invitrogen, BD Biosciences). Even with technological improvements, the general process remains similar.
Although we successfully expressed human colipase, human PTL, rat PTL, rat PLRP1 and rat PLRP2 in the baculovirus system, yields of purified rat and human PLPRP2 were low primarily because the protein precipitated during purification.(8; 13; 15; Lowe, unpublished data)
2.2 Yeast Expression Systems:
For structure-function studies of lipases, an expression system that is easily and quickly manipulated, that processes and secretes lipases correctly and that can produce milligram or larger quantities of proteins is preferable. Yeast expression systems meet these requirements and virtually all labs that produce recombinant lipases utilize yeast, generally the methylotropic yeast Pichia pastoris.(5, 12, 17, 20, 23) Protein expression in P. pastoris is straight-forward and fast.(4) P. pastoris is a single cell organism that is easy to manipulate and grows to high density on inexpensive media. Recombinant mammalian proteins readily undergo correct folding, proteolytic processing, disulfide bond formation and glycosylation. Most vectors contain one of several different yeast signal sequences to target the recombinant protein to the secretory pathway. The vectors take advantage of the ability of P. pastoris to metabolize methanol as its sole carbon source and utilize the strong, methanol-inducible promoter for the alcohol oxidase gene, AOX1, to express the protein of interest. Consequently, the yeast can be grown to high density in glycerol-containing medium without any recombinant protein production. Once the appropriate cell density is reached recombinant protein production is induced by switching to methanol-containing medium. During growth on methanol and medium with no added proteins, P. pastoris secretes low levels of endogenous proteins and the recombinant protein comprises the majority of protein in the medium facilitating subsequent purification.
Expression of pancreatic lipases in P. pastoris
Before starting this expression protocol, clone the cDNA for the lipase of interest in frame in a P. pastoris expression vector. Various vectors have been utilized for pancreatic lipase expression including pHILS1, pPIC9K, pGAPZB, pPICZA and pPICZαA. The last three vectors allow selection of recombinant yeast with zeocin. In our lab, we currently use pPICZαA or pPICZA and have expressed PTL, PLRP2 and CEL from multiple species successfully. The sequences and other properties of these vectors can be found on the Invitrogen website. Currently, Invitrogen offers the PichiaPink expression system. This system utilizes complementation of adenine auxotrophy rather than antibiotic selection to enrich for transformants. The expression of pancreatic lipases using these transfer vectors has not been reported. When pPICZαA vector is used, the nucleotide sequence should be checked to insure the cDNA is inserted in frame with the signal sequence of the vector.
Transformation of P. pastoris
P. pastoris can be transformed by electroporation or chemical methods. We prefer electroporation because it has higher yield of transformants and does not destroy the cell wall of the yeast allowing immediate plating on zeocin selection plates. Electroporation will be described in detail. If you do not have access to an electroporation device, the EasyComp system (Invitrogen) can be used following the manufacturer’s directions.
A. Preparation of transforming DNA
For transformation, the desired plasmid DNA is linearized by restriction digestion. The selection of restriction enzyme depends on the vector and the insert. The restriction enzyme should cut the vector at a single site and not cut the insert. For example, pPICZαA has single Sac I, Pme I and BstX I sites and one of these can be used provided your insert does not contain that restriction site.
Materials:
- Sterile deionized water
- Appropriate restriction enzyme and 10x buffer
- 100x BSA
- 3 M sodium acetate, pH 7.0
- 100% and 80% ethanol
- Optional: Phenol:chloroform:isoamyl alcohol (25:24:1 v/v/v)
- 1.5 ml microcentrifuge tubes
Method:
- Thaw all reagents on ice and briefly centrifuge the tube to get all liquid to the bottom, gently mix the contents. Then mix the following in a 1.5 ml microcentrifuge tube:
- X µl sterile water
- 40 µl 10x restriction buffer (choose the manufacture’s recommended buffer for your enzyme)
- 4.0 µl 100X BSA
- 20 µg of plasmid DNA
- 10.0 µl restriction enzyme (10 U/µl)
Total volume = 400 µl
- Incubate at 37 ºC for 2 hours.
- Add 400 µl of phenol:chloroform:isoamyl alcohol to the digestion reaction mixture, vortex and centrifuge at maximum speed for 1 min in a microcentrifuge. Carefully remove the upper aqueous layer (~360 µl) to another 1.5 ml microcentrifuge tube. Optional: Heat inactivate the enzyme mix at 65 ºC for 20 minutes.
- Add 1/10 volume (~36 µl) of 3 M sodium acetate, mix and add 2.5 volumes (900 µl) of 100% ethanol.
- Incubate on ice for 10 to 15 min and pellet by centrifugation at 15,000 g or higher for 10 min.
- Carefully remove the supernatant. Add 500 µl of 80% ethanol and centrifuge again for 10 min.
- Remove the supernatant and allow the pellet to air-dry. Dissolve the DNA pellet in 10 µl of sterile deionized water. Run 0.5 µl of final DNA solution along with undigested plasmid DNA on 1% agarose gel and stain with Ethidium Bromide to confirm the presence of DNA and complete digestion. Use immediately or store at -20 ºC for later use.
B. Preparation of electrocompetent yeast cells
We generally use GS115 cells for lipase expression. We have not found any advantage to using protease-deficient strains, but they can be used as well (Xiao and Lowe, unpublished results). Use the electrocompetent cells the same day you prepare them. Do not store them.
Materials:
- YPDS plates
- YPD medium and plates
- Ice-cold sterile deionized water (place on ice the day of the electroporation)
- 1 M Sorbitol, sterile and ice cold (place on ice the day of the electroporation)
- 1 litter baffled culture flasks, sterile 50 ml conical tubes, sterile
- 250 ml centrifuge bottles, sterile
Method:
- One week before the intended transformation streak the chosen yeast strain on YPD plate and grow at 30 ºC until single colonies form (~3 days), up to 5 days.
- Two days before the transformation inoculate 3 ml of YPD medium in a sterile 50 ml conical tube with a single colony. Tape the cap loosely on the top of conical tube.
- Incubate at 30 ºC with shaking at 300 rpm overnight.
- Use ~ 100 µl of the overnight grown culture to inoculate 250 ml of YPD medium in a sterile 1 liter baffled flask. Grow the culture at 30 ºC with shaking at 300 rpm for ~ 16 hr. The OD600 of the culture should be 1.3 to 1.5. (It is necessary to dilute the culture before reading the OD to obtain an accurate reading.)
- Divide the culture into two 250 ml centrifuge tubes and centrifuge at 1,500 x g at 4 ºC for 5 min.
- For each centrifuge tube, resuspend the pellet with 125 ml of ice-cold sterile water and centrifuge as above.
- Resuspend the pellet with 50 ml of ice-cold water. Transfer the yeast cells to a 50-ml of conical tube. Centrifuge as in step 5.
- Resuspend the cells with 20 ml of ice-cold 1 M sorbitol. Centrifuge as in step 5.
- Resuspend the cells in 700 µl of ice-cold 1 M sorbitol. Keep on ice and use the same day.
C. Transformation into P. pastoris by Electroporation
Materials:
- 10-12 µg of linearized plasmid DNA in 6 µl of water
- YPDS plates containing 100 µg/ml of zeocin, or varying concentration of zeocin up to 2000 µg/ml if preferred
- 1 M sorbitol, Ice-cold
- Electroporation device such as Bio-Rad GenePulser
- 0.2 cm electroporation cuvettes, chilled on ice
- Glass Pasteur pipettes, sterile
- 2 mm glass beads, sterile
- 15 ml culture tube, sterile
- 0.5 ml microcentrifuge tubes, sterile
Methods:
- Transfer 80 µl of the electrocompetent yeast to a 0.5-ml microcentrifuge tube. Add 6 µl of the linearized DNA. Mix by moving the pipette tip back and forth.
- Incubate on ice for 5 minutes.
- Transfer the mixture to the 0.2 cm chilled cuvette.
- Pulse the cells in the electroporator according to the instrument’s manufacturer’s instructions for yeast.
- Immediately add 0.75 ml of ice-cold 1 M sorbitol to the cuvette. Transfer to a 15 ml culture tube using the sterile glass Pasteur pipette.
- Incubate the culture at 30 ºC with 300 rpm shaking for 2 hours.
- Add 7-10 sterilized glass beads to each of 4 YPDS plates + zeocin. Place 300-350, 200, 100, and 50 µl of the electroporation mix on each plate. Increase the plating volume of electroporation mix with the zeocin concentration if YPDS plates containing varying zeocin concentrations are used. Gently shake the plates horizontally to spread the cells. Let the plates dry for 15 minutes. Remove the glass beads by inverting the plates.
- Incubate the plates upside down at 30 ºC until distinct colonies form in 2-5 days. Occasionally it will take a few days longer.
D. Protein Expression in Pichia pastoris
Materials:
- BMGY medium
- BMMY medium
- 50 ml conical tubes, sterile
- Methanol
Method:
- Inoculate a single colony from the transformation plates into 10 ml BMGY in a sterile 50 ml conical tube. We typically screen around 12 separate colonies for each construct. Incubate at 30 ºC with shaking at 300 rpm for 18-24 hours. The OD600 will be 2-6. Since oxygenation of the culture is important, we typically tape the cap loosely on the top of the conical tube. Alternatively, sterile gauze or PureLink Air Porous Tape can be used to cover the top of the tube.
- Centrifuge at 1,500 x g for 5 minutes. Carefully decant the supernatant and re-suspend the cell pellet in 3 ml of BMMY.
- Return the cultures to the shaking incubator.
- After 12 to 48 hours of methanol induction, withdraw 1 ml of yeast culture and transfer to a 1.5 ml microcentrifuge tube. Pellet the cells by centrifugation at 15,000 x g for 3 minutes. Remove the supernatant and put in a separate tube. Add 1 ml of BMGY to the cell pellet and re-suspend the cells. Store at 4 ºC until the samples are screened for lipase production. We typically can detect robust lipase activity in the media after 12-24 hours and stop our cultures at that point. Alternatively, 100 µl of the culture can be removed at time intervals up to 4 days. If culturing longer we add 0.5% methanol to the culture every 24 hours to maintain induction.
- We screen for lipase production by the standard pH-stat method (See section on lipase assays.) or by protein immunoblot. Alternatively, a fluorescent lipase assay could be employed.
- The yeast with the highest expression levels are selected for storage. Streak the selected yeast transformants onto YPDS plate + zeocin and incubate the plates upside down at 30 ºC until distinct colonies form in 2-5 days.
- To prepare for storage single colonies are cultured in 10 ml YPD medium in a sterile 50 ml conical tube. The culture is incubated at 30 ºC with shaking at 300 rpm until the OD600 of the culture is 2-6, about 18-24 hours. Transfer 800 µl of the culture to a sterile 2 ml cryo-tube. Add 200 µl of sterile glycerol and mix by repeated inversion of the capped tube. The glycerol yeast stock is stored at -80 ºC.
E. Scaling-up Expression
There are two approaches to express larger amounts of recombinant lipase, increased culture volume in large baffled flasks or fermentation. In this section we will only cover growth in baffled flasks since most investigators will not have access to a fermentor and growth in baffled flasks will produce enough recombinant lipase for most purposes. If large amounts of lipase are required for specific experiments, fermentation is generally required. Instructions for fermentation of P. pastoris can be found in literature provided by Invitrogen on their website: http://tools.invitrogen.com/content/sfs/manuals/pichiaferm_prot.pdf
Materials:
- BMGY
- BMMY
- 50 ml conical tube, sterile
- 1 or 2 liter baffled flasks with porous caps, sterile
- Large capacity centrifuge bottles, sterile
- Methanol
Method:
- Inoculate a single colony into 5 ml of BMGY in a 50 ml conical tube. Grow at 30 ºC overnight in a shaking incubator at 300 rpm.
- Inoculate 500 ml of BMGY in each of two 2-liter baffled flasks with half of the culture from step 1. Grow as above until an OD600 of 2-6 is reached.
- Harvest the cells by centrifugation in sterile centrifuge bottles at 1,500 x g for 5 minutes at room temperature. Decant the supernatant and add 200 ml of BMMY to re-suspend the cell pellet.
- Put the resuspended yeast in fresh sterile baffled flasks. The culture can be divided into 100 ml in each of two 1 liter flasks or 200 ml in a 2 L flask. Return to the incubator and continue to grow with shaking at 300 rpm.
- Add 100% methanol to 1.0 % every 24 hours. When expressing a new lipase or construct we typically remove aliquots of the culture at increasing time points up to 4 days to determine the optimal time of induction. We prefer to do the time course on the scaled-up expression rather than on the small scale expression because the optimal time for induction frequently differs between the small and large-scale expression cultures.
- Harvest the cells and supernatant by centrifugation at 3, 000 x g for 5 minutes at room temperature. Recombinant pancreatic lipases will be secreted into the medium. We generally proceed directly to purification at this point. The medium can be stored at -80 ºC and processed later.
3. Recombinant Lipase Protein Purification
3.1 Purification of Recombinant PTL and PLRP2 Proteins
We routinely purify recombinant PTL and PLRP2 on 1-ml Mono S FPLC (Pharmacia Biotech) or 5/50 GL (GE Healthcare) column using Chromatography system. The one-step purification usually yields good outcome. A second step purification by gel filtration could be followed if further purification is needed. Below is the detailed procedure for purifying human PTL. The purification of PTL from other species or PLRP2 can be carried out by following this protocol with minor modifications. The modifications generally involve the adjustment of the pH of Binding/Washing or Elution buffer according to the theoretical isoelectrical point (IP) of the protein of interest. The principle is that the pH for the buffers employed in this procedure is 0.3-0.5 below the IP of the protein of interest.
Materials:
- 200 mM MES, pH 6.5
- 10 N NaOH or HCl
- Binding/Washing buffer: 10 mM MES, pH 6.5
- Elution buffer: 10 mM MES, 200 mM NaCl, pH 6.5
- 500 ml 0.22 µm low protein binding Bottle Top Filter(Cat# 431118, Corning Incorporated, Corning, NY)
- 25 mm 0.22 µm PVDF syringe filters
- Amicon Ultra Ultracel-30K (Cat# UFC903024, Millipore, )
- Dialysis tubing cellulose membrane, flat width 31 mm, 3.1 ml/cm, MWCO: 20 kDa (Cat# 131348, Spectrum Laboratories, Rancho Dominguez)
- 1 ml Mono S FPLC (Cat#9620034, Pharmacia Biotech) or Mono S 5/50 GL (Cat#17-5168-01, GE Healthcare, Piscataway, NJ)
- AKTA explorer chromatography system(Amersham)
- 13x100 mm glass culture tubes, clean
Method:
- Centrifuge the supernatant (~ 200ml) at 10,000g for 10 min at 4 ºC. The resulting supernatant is further clarified by filtering through 0.22 µm cup filter.
- Concentrate the supernatant down to ~ 30 ml using preferred system with the membrane MWCO of 10-30 kDa.
- In a dialysis tube with the membrane MWCO of ~ 20 kDa, dialyze the concentrated protein against 1-2 liter of ddH2O with 2 mM benzamine overnight in the cold room.
- Adjust the protein sample with 200 mM MES and 10 N NaOH to 10 mM MES, pH 6.5.
- Filter the protein sample again with 0.22 µm syringe filter.
- After equilibrating the Mono S column with Bind/Washing buffer, load the prepared protein sample with the flow rate of 1 ml/min.
- Wash the column thoroughly with ~ 25 ml of Binding/Washing buffer.
- Elute the bound protein with increasing concentrations of NaCl up to 200 mM within 60 min. The bound proteins usually come off the column between 60-120 mM NaCl.
- The concentrations of eluted protein are monitored by A280 reading. The fractions represent the protein elution peaks were collected and further evaluated by both lipase activity assay and SDS-PAGE gel staining assay.
- Pool the desired fractions together. Concentrate and exchange with buffer to 20 mM Tris-HCl, pH 8.0 with Amicon Ultra Ultracel-30K.
- Determine the protein concentration with A280, with the extinction coefficiency Abs 0.1% =1.246. The total yield usually ranges from 2-8 mg. Store aliquots of the final purified protein at -80 ºC for later use.
3.2 Purification of the recombinant CEL Protein
We routinely purify the recombinant CEL protein using Heparin affinity column coupled with AKTA Explorer chromatography monitor system (Amersham).
Materials:
- Binding/washing buffer: 20 mM sodium phosphate, 1 mM EDTA, pH 7.4
- Elution buffer: 20 mM sodium phosphate, 1 mM EDTA, 1 M NaCl, pH 7.4
- 10 N NaOH
- 25 mM sodium phosphate, 150 mM NaCl, pH 7.4
- 500 ml 0.22 µm low protein binding Bottle Top Filter(Cat# 431118, Corning Incorporated, Corning, NY)
- 25 mm 0.22 µm PVDF syringe filters
- Dialysis tubing cellulose membrane, flat width 31 mm, 3.1 ml/cm, MWCO: 20 kDa (Cat# 131348, Spectrum Laboratories, Rancho Dominguez)
- Dialysis tubing cellulose membrane, flat width 76 mm, 21 ml/cm, MWCO:12 kDa (Cat# D9402, Sigma, St. Louis, MO)
- Dialysis tubing cellulose membrane, flat width 31 mm, 3.1 ml/cm, MWCO: 20 kDa (Cat# 131348, Spectrum Laboratories, Rancho Dominguez)
- 5 ml HiTrap Heparin HP (Cat# 17-0407-01, GE Healthcare, Piscataway, NJ)
- AKTA explorer chromatography system(Amersham)
Method:
- Centrifuge the supernatant (~ 200ml) at 10,000g for 10 min at 4 ºC. The resulting supernatant was further clarified by filtering through 0.22 µm Bottle Top Filter.
- Dialyze the clarified supernatant with a large dialysis tube (MWCO:12 kDa) in 4 liter ddH2O with 2 mM benzamidine overnight in the cold room. Caution-We experienced substantial loss of CEL protein during concentration, therefore protein concentrating is not recommended.
- Use 1M sodium phosphate, 500 mM EDTA, and 10 N NaOH to adjust the protein sample to a final concentration of 20 mM sodium phosphate, pH 7.4.. Alternately, dilute the supernatant with equal volume of ddH2O, then adjust pH to 7.4 with 10 N NaOH.
- Filter the protein sample again with 500 ml Bottle top Filter (0.22 µm) filter.
- After equilibrating the 5-ml Heparin column with 50 ml Binding/Washing buffer, load the prepared protein sample with the flow rate of 2.5-5 ml/min, which is monitored by AKTA explorer chromatography system.
- Wash the column thoroughly with ~ 100 ml of Binding/Washing buffer.
- Elute the recombinant protein with increasing concentration of NaCl up to 800 mM within 60 min, with the flow rate of 1-2 ml/min. The bound protein usually elutes from the column between 300-600 mM NaCl.
- The concentrations of eluted protein are monitored by A280 reading. The fractions represent the protein elution peaks were collected and further evaluated by both lipase activity assay and SDS-PAGE gel staining assay.
- Pool the desired fractions together. Dialyze the pooled protein samples against 1 liter of 25 mM sodium phosphate, 150 mM NaCl, pH 7.4 overnight at the cold room in a dialysis tube (3.1 ml/cm, MWCO: 20 kDa). Determine the protein concentration with A280, with the extinction coefficiency Abs 0.1% =1.341. The total yield usually ranges from 3-6 mg. Aliquots the final purified protein and stored at -80 ºC for later use.
Figure 1 shows the analysis of the purified PTL, PLRP2 and CEL by SDS-PAGE. Lanes A and B demonstrate the low level of proteins secreted by P. pastoris. The predominant band in the medium is the recombinant lipase.

Figure 1. Expression of recombinant pancreatic lipases. GelCode blue stained acrylamide gels are shown. Panels A & B: media from PTL-expressing P. pastoris cultures. C. PTL expressed and purified from baculovirus infected insect cells. D. PTL expressed and purified from P. pastoris. E. PLRP2 expressed and purified from baculovirus infected insect cells. F. PLRP2 expressed and purified from P. pastoris. G. CEL expressed and purified from P. pastoris.
Recipes:
20% Dextrose (10X):
Dissolve 200 grams of about 800 ml of distilled water and bring to a final volume of 1000 ml. Autoclave for 15 minutes or filter sterilize. Store at room temperature.
YPD Medium:
To prepare 1000 ml dissolve 10 g yeast extract and 20 g peptone in 900 ml of water. Autoclave for 20 minutes on liquid cycle. Let the autoclaved solution cool to less than 60 ºC and add 100 ml of the 20% dextrose.
YPDS medium:
Prepare as above and add 91.1 g sorbitol to the medium prior to autoclaving.
YPD agar plates:
To prepare 500 ml of YPD agar dissolve 5 g yeast extract and 10 g of peptone in 450 ml of water. Add 10 g of agar and a magnetic stir bar. Autoclave for 20 minutes on the liquid cycle. Let the solution cool to less than 60 ºC and add 50 ml of 20% dextrose. If zeocin plates are required, add 500 µl from a 100 mg.ml zeocin stock solution to the cooled agar solution. Add the zeocin while stirring on a magnetic stir plate. Let stir for several minutes after adding to insure complete mixing. Pour the media into 100 mm Petri dishes and let dry on the bench overnight. If zeocin is included protect the plates from light by covering with black plastic. Store at 4 ºC. The shelf life is 1-2 weeks.
YPDS agar plates:
Prepare as above and add 91.1 g sorbitol to the medium prior to autoclaving.
BMGY:
Dissolve 10 g of yeast extract and 20 g peptone in 700 ml of water and autoclave for 20 minutes on the liquid cycle. Cool to room temperature and add:
- 100 ml 1M potassium phosphate, pH 6.0 (combine 24 ml of K2HPO4 and 156 ml of 1 M KH2PO4 and confirm the pH is 6.0 adjust with phosphoric acid or KOH if required);
- 100 ml 10 X YNB (Dissolve 134 g of yeast nitrogen base (YNB) with ammonium sulfate and without amino acids in 1000 ml by gently heating the solution until the YNB dissolves. Filter sterilize. Store at 4 ºC);
- 20 ml 50 x biotin (Dissolve 20 mg biotin in 100 ml of water and filter sterilize. Store at 4 ºC);
- 100 ml 10 X glycerol (100 ml glycerol and 900 ml of water. Mix well and filter sterilize. Store at room temperature.)
Store medium at 4 ºC. The shelf life is about two months.
BMMY:
Combine the ingredients for BMGY. Add 100 ml of 10 x methanol (5 ml of methanol with 95 ml of water.) Filter sterilize.
4. Assays for Pancreatic Lipases
Lipase assays are performed routinely in clinical laboratories and in research laboratories with an interest in lipases. The assay requirements vary greatly depending on the setting and the use. The clinical laboratory requires specific, sensitive, quick and reproducible automated assays. In the research lab, investigators interested in the biochemistry or structure-function analysis of lipases need specific, sensitive and reproducible assays that allow them flexibility in changing the assay conditions. Investigators interested in molecular evolution of lipases or in identifying small molecule inhibitors of lipases require automated assays suitable to screening multiple samples oftentimes in multi-well plates. Consequently, many different lipase assays are available. The numerous methods can be classified as: a) titrimetric; b) spectrometric (photometric, fluorimetric, infra-red); c) radiometric; d) turbidometric; e) chromatographic; f) interfacial tensiometry; and g) conductimetric. A general discussion of these methods can be found in a recent review and this section will focus on several assays that are primarily suitable for the research laboratory.(2,9)
4.1 Lipase Substrates
Either triglycerides or diglycerides are predominantly utilized in lipase assays. Diglycerides are mainly employed in coupled enzyme assays that produce colored or fluorescent products or in assays of monolayers in the Wilhemy plate method using interfacial tensiometry. Diglycerides do have several drawbacks. They are less stable and more expensive than triglycerides and can be hydrolyzed by nonspecific esterases. In most assays, triglycerides are the preferred substrate. All lipases have activity against a broad range of triglycerides containing short, medium or long-acyl chains. Among the available triglycerides tributyrin, trioctanoin and triolein are the most commonly used substrates. All three are liquid at normal assay temperatures. Triolein is highly specific for lipases since no other esterase is able to hydrolyze long-chain acylglycerols at an oil-water interface. Because pancreatic lipases have the highest specific activity against tributyrin and tributyrin is the least expensive of the three substrates, it is frequently used to quantitate lipase activity. The downside is that esterases other than lipases can hydrolyze tributyrin. If tributyrin is employed to detect lipases in tissue extracts or partially purified lipases, the presence of a true lipase must be confirmed by demonstrating activity against triolein.
Other substrates have been proposed for lipase assays. Several groups have developed assays with fluorescent substrates.(1,16) These assays are fast and can be done with readily available equipment. The major drawback is that the substrates are sensitive to oxidation by atmospheric oxygen, a problem that precludes use in routine conditions. An ultraviolet spectrophotometric assay using long-chain triglycerides from Aleurites fordii seeds has been described and may be useful for screening multiple samples for the presence of lipase.(14) The main limitation is that the substrate is not readily available.
4.2 pH-stat Titrimetric Assay
Continuous titration with a pH-stat coupled to an automatic burette and recorder is a versatile method. It allows for investigations of lipase kinetics and permits the investigator to vary the assay conditions, including substrates and bile salts. Because the method depends on titration of the released fatty acid, pH affects the sensitivity considerably and pH effects on activity cannot be determined in the pH-stat. In addition, the equipment is not readily available in most laboratories.
Materials:
- A titrator consisting of an autoburette, a micro-electrode, propeller stirrer, thermostatted reaction vessel connected to a circulating water bath and computer to run the titration program, record results and perform calculations of activity. Alternatively, heating tape and controller can be used to heat a standard laboratory beaker. The reaction vessel should be glass. Both lipases and colipase bind to plastic reaction vessels and they should not be used. A propeller stirrer is more efficient than a magnetic stirrer and is preferred. Suitable equipment can be obtained from Radiometer (Copenhagen, Denmark) or Mettler (Ohio, USA).
- Lipase substrates usually tributyrin, trioctanoin or triolein. The choice of substrate depends on the requirements of the research. Tributyrin is less expensive than the other substrates. Since most lipases have higher specific activity against tributyrin, it is useful for screening expression levels or for monitoring purification. Long-chain triglycerides are the natural substrate for pancreatic lipases and triolein provides a reasonable choice when characterizing the properties of a neutral lipase. Additionally, triolein is only cleaved by lipases and not by other esterases as can be seen with tributyrin. Store substrates at 4 ºC.
- Assay buffer: 1mM Tris-HCl, pH 8.0, 0.15 M NaCl, 2.0 mM CaCl2, ±4 mM sodium taurodeoxycholate. For assays of CEL, we also add 3.75% de-fatted BSA and use 10 mM tauroceoxyholic acid instead of sodium taurodeoxycholate. Can be stored at room temperature.
- Dounce homogenizer, blender or bath sonicator
- Titrant: 50 or 100 mM NaOH
- For pancreatic triglyceride lipase (PTL) assays: Colipase stock solution of 1 mg/ml dissolved in 10 mM Tris-HCl, pH 8.0 and 0.15 M NaCl. Porcine colipase can be obtained from Sigma. Background activity should be checked because commercially obtained colipase can be contaminated with lipase. Store in aliquots at – 80 ºC. To avoid repeated freeze/thaw cycles, the working stock can be stored at 4 ºC for several weeks.
Assay:
- Prepare the substrate solution by mixing 0.5 ml of substrate (0.1 ml of triolein can be used) with 14.5 ml of assay buffer for each reaction. It is best to prepare the amount of substrate required for the number of planned assays. Emulsify the mixture with 6-10 strokes in a Dounce homogenizer, in a blender or in a bath sonicator or with a probe sonicator. The emulsion should be homogenous and is stable for 4-6 hours.
- Turn on the pH-stat and the circulating water bath or temperature controller. Flush the burette according to the manufacturer’s directions. Set the circulating water bath to 37 ºC.
- Add 15 ml of the substrate emulsion to a 20 ml glass beaker or a similar size thermal jacketed vessel. If using a beaker, wrap it with heating tape according to the manufacture’ directions. Place the pH electrode, glass delivery tip and propeller stirrer in the substrate solution. It is important to stir the solution rapidly. Set the propeller stirrer to the highest speed that does not produce bubbles in the substrate solution.
- If required, add colipase to the reaction. To measure PTL activity a 2-5 fold molar excess of colipase over lipase is sufficient. If the concentration of lipase is not known, then activity with several concentrations of colipase should be determined. If colipase is in excess the activity should increase minimally.
- Read just the pH of the substrate solution to pH 8.0 with 1 N NaOH or 1 N HCl. In general, NaOH is required. The titrator program can be set up to allow a run-in period for the pH adjustment.
- Add the lipase sample and start monitoring the reaction. Up to 1 ml of sample volume can be added although high protein concentrations may inhibit lipase activity. Detectable and reproducible reaction rates can be seen with around 50 ng of PTL. Larger amounts of PLRP2 and CEL are required since they have lower activities than pancreatic triglyceride lipase.
- Typically, reactions are run for 5 to 10 minutes and the initial rate calculated from the linear portion of the curve. The program for the titrator will calculate the rate based on the titrant concentration and the mean slope of the reaction. Specific activities are determined by dividing the result by the lipase concentration. The results are expressed as International Units (IU)/mg where 1 IU equals 1 µmole of fatty acid released/min. If unknowns are being measured, the amount of lipase present can be estimated from the known specific activity of the lipase against the substrate employed.
Figure 2 shows a typical assay result of PTL with and without colipase using the pH-stat method. The Table includes activities of purified PTL, PLRP2 and CEL against three different substrates.

Figure 2. pH-stat assay of PTL activity against tributyrin. Two µg of PTL was added to a tributyrin emulsion in 4 mM NaTDC as described. After 5 min, 2 µg of colipase was added. The reaction velocity before and after colipase addition can be calculated from the slope of each line.
Table: Specific Activities of Pancreatic Lipases
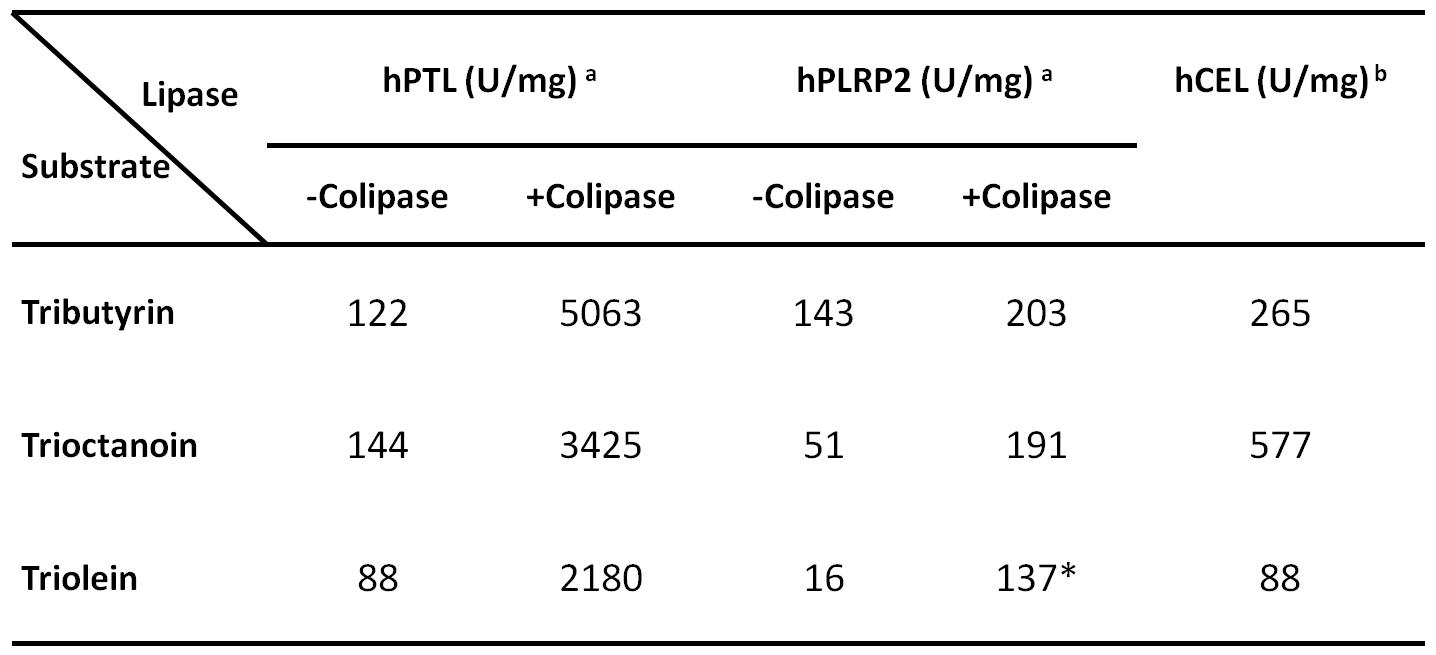
a Assayed in 4 mM NaTDC with or without 5 molar excess colipase at 22 ºC
b Assayed in 12 mM NaC
* Achieved after ~ 17.5 min lag time or with the addition of oleic acids
4.3 Radiometric Assay
The release of radiolabeled fatty acids from triacylglcerides is a sensitive method that is applicable in many situations.(7, 11) The released fatty acids are separated from the remaining diglycerides and triglycerides by paper- or thin-layer chromatography or by phase partition extraction. The method is discontinuous and can give misleading results if the assay is not stopped while the reaction velocity is still linear. An important limitation of this method is that the range of commercially available radioactive substrates is limited. Thus, it is hard to screen for activity against multiple substrates.
Materials:
- Triolein: both nonradioactive and radioactive triolein are required. Usually 3H-triolein is used.
- Assay Buffer: 30 mM Tris-HCl, pH 8.0, 1.0 mM CaCl2, ± 4 mM taurodeoxycholate. For assays of CEL use 10 mM taurocholic acid and add 3.75% de-fatted BSA.
- Colipase stock solution for assays of PTL (See above)
- Extraction Solvent: chloroform/methanol/heptane (12.5:14.0:10.0)
- Carbonate Solution: 50 mM sodium carbonate, pH 10.5
Assay:
- Add 2.0 µl of triolein (1.56 µmoles) to 5.0 ml of assay buffer in a glass test tube and vortex for 2 minutes. The final concentration of triolein is 0.312 mM.
- Transfer enough substrate solution for the number of assays planned to a glass tube. Allow 50 µl for each assay. Add 30 µl of 3H-triolein to each ml of assay mix. Sonicate for 5 minutes in a bath sonicator or vortex for several minutes. The resulting emulsion will be milky white and will have a specific activity of 333 dpm/ml of fatty acid.
- If needed dilute the colipase stock 1:10 in assay buffer.
- Mix 45 µl of substrate with 2.5 µl of colipase as required and with 2.5 µl of the lipase sample. A glass tube should be used.
- Vortex the mixture briefly and incubate at room temperature for the desired time. Five to 10 minutes is usually adequate. It is best to determine the activity at several time points to insure that the reaction is linear. Always include an assay blank where buffer is substituted for the lipase.
- Stop the reaction by adding 750 µl of the extraction solvent. Vortex briefly and add 250 µl of the sodium carbonate solution. Vortex vigorously to completely mix the organic and aqueous phases.
- Centrifuge at room temperature for 5 minutes at top speed in a table-top centrifuge. A clear distinction between the upper aqueous phase and the lower organic phase will be present. Often a white precipitate will be present at the interface and care should be taken not to disturb the layer when sampling the upper phase.
- Remove 100 µl from the top phase and count in a scintillation counter.
- Calculate the results using the following formula:
- (dpm counted) x (total volume in the top phase)
- (3333) x (volume of top phase counted) x (assay time in minutes)
- This equation assumes that all of the free fatty acids are contained in the top phase. In fact, about 75% of the oleic acid partitions to the top phase. The exact percentage can be determined by adding radiolabeled oleic acid to the triolein instead of radioactive triolein. Phase extract the mixture as above and count 100 µl of the top phase and use the total volume of the top phase to calculate the percentage of oleic acid remaining in the top phase. Prior to centrifugation remove an aliquot of the freshly vortexed mixture and count in scintillation fluid to determine the total radioactivity in the mix. Use these two values to determine the efficiency of oleic acid extraction and use this number to correct experimental results.
5. References
- Beisson F, Ferte N, Nari J, Noat G, Arondel V, Verger R. Use of naturally fluorescent triacylglycerols from Parinari glaberrimum to detect low lipase activities from Arabidopsis thaliana seedlings. J Lipid Res. 40: 2313-21, 1999. PMID: 10588957
- Beisson F, Tiss A, Riviere C, Verger R. Methods for lipase detection and assay: a critical review. European Journal of Lipid Science and Technology. 102: 133-53, 2000.
- Berton A, Sebban-Kreuzer C, Rouvellac S, Lopez C, Crenon I. Individual and combined action of pancreatic lipase and pancreatic lipase-related proteins 1 and 2 on native versus homogenized milk fat globules. Mol Nutr Food Res. 53: 1592-602, 2009. PMID: 19824014
- Daly R, Hearn MT. Expression of heterologous proteins in Pichia pastoris: a useful experimental tool in protein engineering and production. J Mol Recognit. 18: 119-38, 2005. PMID: 15565717
- Eydoux C, De Caro J, Ferrato F, Boullanger P, Lafont D, Laugier R, Carriere F, De Caro A. Further biochemical characterization of human pancreatic lipase-related protein 2 expressed in yeast cells. J Lipid Res. 48: 1539-49, 2007. PMID: 17401110
- Hui DY, Howles PN. Carboxyl ester lipase: structure-function relationship and physiological role in lipoprotein metabolism and atherosclerosis. J Lipid Res. 43: 2017-30, 2002. PMID: 12454261
- Kaplan A. A simple radioactive assay for triglyceride lipase. Anal Biochem. 33: 218-25, 1970. PMID: 5445316
- Lowe ME. Human pancreatic procolipase expressed in insect cells: purification and characterization. Protein Expr Purif. 5: 583-6, 1994. PMID: 7858428
- Lowe ME. Assays for pancreatic triglyceride lipase and colipase. In: Doolittle MH, Reue K, editors. Methods in Molecular Biology: Lipase and Phospholipase Protocols. Totowa NJ: Humana Press Inc. p. 59-70, 1998.
- Lowe ME. The triglyceride lipases of the pancreas. J Lipid Res. 43: 2007-16, 2002. PMID: 12454260
- Marsh DG, George JM. Importance of sulfhydryl groups for lipolytic action in the isolated fat cell. J Biol Chem. 244: 1381-2, 1969. PMID: 4304193
- Murasugi A, Asami Y, Mera-Kikuchi Y. Production of recombinant human bile salt-stimulated lipase in Pichia pastoris. Protein Expr Purif. 23: 282-8, 2001. PMID: 11676603
- Payne RM, Sims HF, Jennens ML, Lowe ME. Rat pancreatic lipase and two related proteins: enzymatic properties and mRNA expression during development. Am J Physiol. 266: G914-G21, 1994. PMID: 8203536
- Pencreac'h G, Graille J, Pina M, Verger R. An ultraviolet spectrophotometric assay for measuring lipase activity using long-chain triacyglycerols from Aleurites fordii seeds. Anal Biochem. 303: 17-24, 2002. PMID: 11906146
- Roussel A, Yang Y, Ferrato F, Verger R, Cambillau C, Lowe M. Structure and activity of rat pancreatic lipase-related protein 2. J Biol Chem. 273: 32121-8. PMID: 9822688
- Salvayre R, Negre, A, Radom, J., and Douste-Blazy, L. Fluorometric Assay for Pancreatic Lipase. Clin Chem. 32: 1532-6, 1986. PMID: 3731446
- Sebban-Kreuzer C, Deprez-Beauclair P, Berton A, Crenon I. High-level expression of nonglycosylated human pancreatic lipase-related protein 2 in Pichia pastoris. Protein Expr Purif. 49: 284-91, 2006. PMID: 16861001
- Thirstrup K, Carriere F, Hjorth S, Rasmussen PB, Woldike H, Nielsen PF, Thim L. One-step purification and characterization of human pancreatic lipase expressed in insect cells. FEBS Lett. 327: 79-84, 1993. PMID: 8335100
- Thirstrup K, Carriere F, Hjorth SA, Rasmussen PB, Nielsen PF, Ladefoged C, Thim L, Boel E. Cloning and expression in insect cells of two pancreatic lipases and a procolipase from Myocastor coypus. Eur J Biochem. 227: 186-93, 1995. PMID: 7851384
- Trimble RB, Lubowski C, Hauer CR, 3rd, Stack R, McNaughton L, Gemmill TR, Kumar SA. Characterization of N- and O-linked glycosylation of recombinant human bile salt-stimulated lipase secreted by Pichia pastoris. Glycobiology. 14: 265-74, 2004. PMID: 14693913
- van Oers MM. Opportunities and challenges for the baculovirus expression system. J Invertebr Pathol. 107 Suppl: S3-15, 2011. PMID: 21784228
- Whitcomb DC, Lowe ME. Human pancreatic digestive enzymes. Dig Dis Sci. 52: 1-17, 2007. PMID: 17205399
- Yang Y, Lowe ME. Human pancreatic triglyceride lipase expressed in yeast cells: purification and characterization. Protein Expr Purif. 13: 36-40, 1998. PMID: 9631512