
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2018.15
| Attachment | Size |
|---|---|
| 728.16 KB |
1. Introduction
In 1938, Dr. Dorothy Anderson published a paper describing the characteristic of Cystic Fibrosis (CF) in the pancreas; the term of “cystic fibrosis” refers to the autopsy findings of fibrosis with cyst in the pancreas of children who died early in life with this disease. In 1949 Anderson also discovered that CF is a genetic condition. More recently this was described to be caused by mutations in the CF transmembrane conductance regulator (CFTR) (83). Other than the pancreas, CF also affects the lungs, liver, kidneys, male reproductive and gastrointestinal tract (4, 82). The disease leads to shortened life expectancy most often due to respiratory failure resulting from airway obstruction, bacterial infections and inflammation (9, 98).
CFTR, a member of the ATP-binding cassette (ABC) transporter protein family, is the cAMP-dependent Cl channel at the apical membranes of most epithelial cells (75). Mutations of CFTR gene cause CF, which is the most common fatal autosomal recessive disorder with a disease frequency of 1 in 2,500 live births and a carrier rate of approximately 5% in Caucasian population (33, 114). The disease is characterized by a malfunction of exocrine tissues due to dysregulation of an epithelial chloride (Cl) channel. The major clinical features include chronic pulmonary disease, pancreatic exocrine insufficiency, intestinal disease (especially constipation) and an increase in the concentration of sweat chloride (87). In the lungs, airways become colonized with bacteria and repeated pulmonary infections ensue. The recurrent infections and inflammation result in submucosal gland hypertrophy and excessive mucus secretion. The impaired mucociliary clearance and plugging of small airways cause progressive bronchiectasis and ultimately lead to respiratory failure (100). Following the lung, the pancreas is the most affected organ in CF. It has been documented that most of the CF patients have pancreatic exocrine insufficiency, which leads to maldigestion and potentially malnutrition (117). In this context, malabsorption of fat and fat-soluble vitamins are the most common nutritional deficient seen in this disease.
In this chapter, we will focus on how decreased CFTR function leads to protein plugging of the ducts and pancreatic atrophy. We will also shed light on the latest animal models to better understand the CF pancreatic disease and its relationship to chronic pulmonary disease and intestinal disease.
2. CF Animal Models
Mice, rats, pigs, and ferrets for CF research
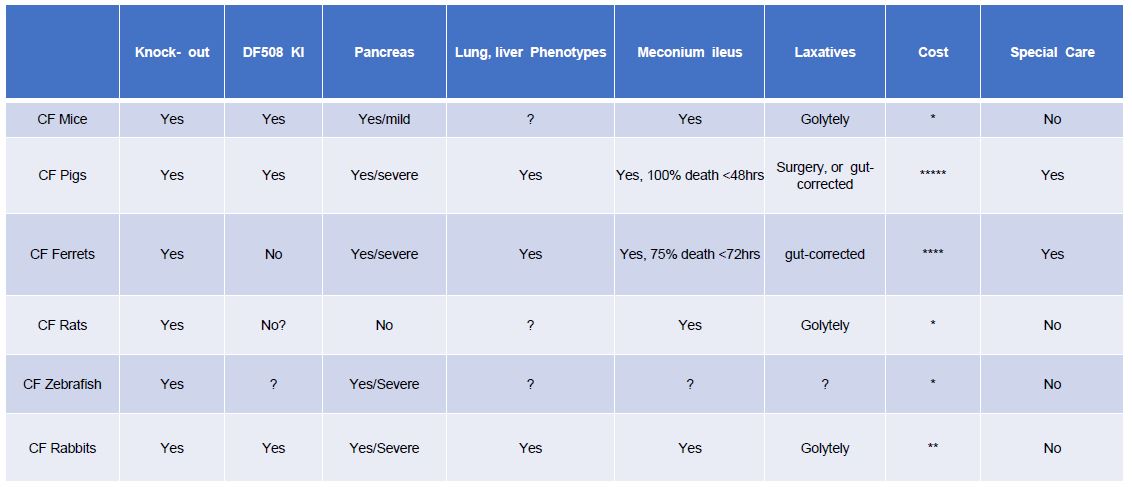
Since the discovery of the CF gene many animal models have been generated to mimic the CF symptoms in human patients. The earliest models were in mice with ∆F508 mutation -cftr mutation (16, 89, 96, 116). CF mouse models have made significant contributions toward our understanding of the disease and the development of therapies (96). Different CF mouse models have been developed, such as the exon 10 knockout (KO) model, the ∆F508 model (16, 105), and the G551D model (22). However, significant limitations have been acknowledged in translating the information gained from CF mice to the humans. For example, unlike human CF patients, CF mice show neither pulmonary pathophysiology nor obvious pancreatic pathology or liver problems (88). Recently established CFTR KO rats recapitulate several features of human CF disease; however, they do not develop spontaneous lung infections (96). CFTR KO ferrets, and CFTR KO and ∆F508 pigs generated by nuclear transfer have shown a similar pathology to that observed in human CF patients (101), including lung, pancreatic and liver phenotypes that were not often found in CF mice. However, neither pigs nor ferrets are convenient laboratory species. Both CF ferrets and CF pigs suffer from meconium ileus, which causes these animals to die within a few days after birth; therefore, they are associated with high maintenance cost and require special animal handling skills (45). These factors have limited the applicability of CF pigs and ferrets almost exclusively to the labs originally producing these animals and a few groups closely associated with them (Table 1). An ideal animal CF model would mimic the characteristics of human CF patients including the pancreatic insufficiency, but not require exceptional expertise or resources.
Table 1. Characteristics of cystic fibrosis and key clinical consequences noted in animal models. With emphasis on the rabbit model the table summarizes the differences of phenotypes of CF in all known animal models. In addition, we showed the type of laxatives that will help overcome the gut impaction. The pancreas defect was also shown in the different models. * represents the cost of the animal model on a 1 to 5 scale.
Rabbits for CF research
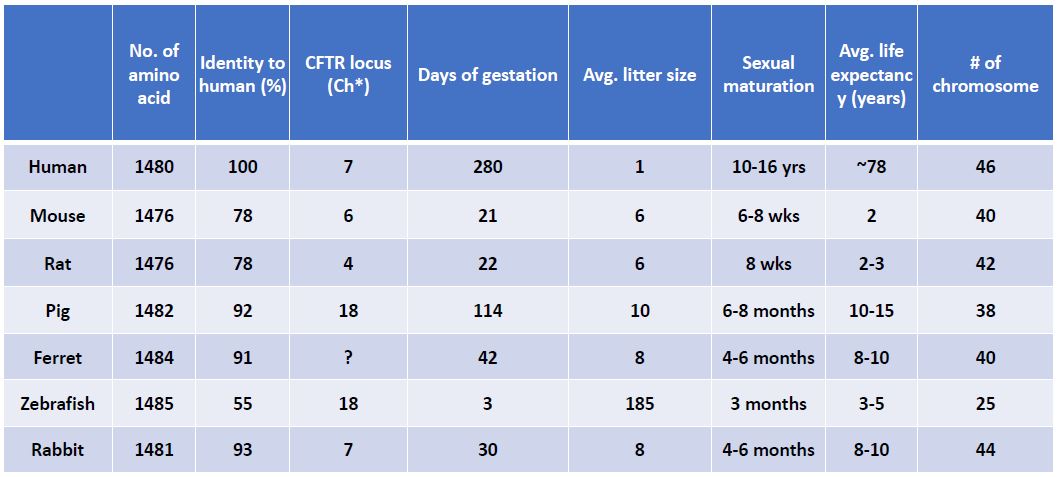
There are anatomical, genetic, and biochemical similarities between rabbit and human (44), making the rabbit a potentially more relevant model for biochemical, molecular and physiological characterization of CF pathology and for the development of CF therapies than mice. As shown in Table 2, the amino acid sequence of rabbit CFTR shares about 93% identity to that in human (Table 2). Rabbit also has a chromosome arrangement that is similar to humans: 44 chromosomes in rabbit vs. 46 in human; both rabbit and human CFTR genes are present on chromosome 7. Compared to other large animals, such as the pig and ferret, a rabbit is a standard lab animal species that can be easily housed in most research institutes and is relatively economically affordable. Though rabbits airways have some anatomic and physiologic features similarly found in humans, the main concern associated with using rabbits for CF research is the absence of airway submucosal glands (SMGs) in rabbits (115). Since CFTR is abundantly expressed in SMGs of human airways, it has been hypothesized that dysfunction of SMGs initiates CF-like lung disease, leading to mucus accumulation observed in CF patients, as well as in CF pigs and ferrets, both of which contain SMGs in their airways. On the other hand, the absence the glands from mice airways has been cited as one of the explanations for lack of lung disease in CFTR KO mice. Therefore, it has been predicted that CFTR deficient/defective rabbits are unlikely to display mucociliary defects and spontaneous lung infections associated with CF. However, our preliminary data reveals that CF rabbits have a similar airway pathology to that in human CF patients (Tables 1 & 2). Although CFTR-/- KO rabbits eventually develop distal intestinal obstruction, meconium ileus is rarely observed in the animal within the first month after birth because rabbit has a large functional cecum. To some extent, our work challenges the traditional view on the importance of SMGs in CF pathology, suggesting that SMGs may not be a critical player for the development of CF lung disease. In support of this view, overexpression of βENaC in mice can produce mucus obstruction in the small, non-glandular airways (57), which are thought to be the site of disease initiation in cystic fibrosis neonates (124). In CF pigs, the mucus appears to arise from goblet cells in the surface of the epithelium of the airways (99). Mucus accumulation in CF ferret airways is associated with variable levels of goblet and mucus cell hyperplasia in the surface airway epithelium and submucosal glands (101). More recent data indicated that defective goblet cell exocytosis in CFTR KO mice contributes to CF-associated disease in the intestine (55). In fact, though SMGs in airways might be a primary site for CF pathogenesis, a critical role for mucus-producing goblet cells in CF airway pathology has not been excluded. Indeed, our preliminary data revealed that the goblet cells, which exist in rabbit airways, maybe the primary contributor to mucus accumulation in the airways of CFTR KO rabbits(Table 1 & 2).
Table 2. CFTR related characteristics among species.
ΔF508 mutation
Mutations of the gene encoding CFTR lead to CF, and more than 2023 CF mutations (disease related or not) have been identified (http://www.genletsickkids.on.ca/cftr/) in the CFTR gene. The most common mutation in CF is the deletion of the phenylalanine residue at position 508 (ΔF508) (13), which is in the first nucleotide binding domain of CFTR. The ΔF508 mutation is present in more than 90% CF patients. A critical issue in CF disease is the inability of ΔF508CFTR to achieve the native, folded state required for its export from the endoplasmic reticulum (ER) and traffic to the cell surface. Instead, ΔF508 protein is exclusively retained in the ER and degraded by the ubiquitin-proteasome system (13, 43, 113). A therapeutic strategy aimed at facilitating ΔF508 folding and trafficking is highly desirable for treatment of the disease because ΔF508 mutant has a substantial CFTR Cl current if it reaches the cell surface (23). A new drug, Orkambi, that combines a CF corrector (Lumacaftor, which acts as a chaperone for a correct protein folding which increases the number of CFTR proteins to the cell surface) and potentiator (Ivacaftor, increases the activity of the CFTR (conductance) at the cell surface) recently has received breakthrough therapy designation to treat CF patients with ΔF508CFTR. However, this drug only improves lung function assayed by forced expiratory volume in 1 second (FEV1) by 2.6-4%. Therefore, further research on ΔF508 mutation is needed to develop a better drug to treat CF patients.
3. CFTR Deficiency in CF
The Cftr gene encodes the CFTR protein, a member of the ABC transporter superfamily, which is the cAMP-dependent Cl channel at the apical membranes of most epithelial cells, making it unique among members of this protein family (75). The CFTR protein migrates to the surface of cells that line the pancreatic duct, airways, gastrointestinal tract, biliary tract, part of the male reproductive tract and cells that are part of sweat glands (50, 75, 79). CFTR forms a pore or channel that allows ions, including chloride and bicarbonate, to move from one side of the cell membrane to the other (Figure 1) (58, 83). Channel activation is mediated by cycles of regulatory (R) domain phosphorylation by PKA/PKC, ATP-binding to the nucleotide-binding domains, and ATP hydrolysis (Figure 1). Demonstration that CFTR functions as a chloride channel regulated by cyclic AMP (cAMP)-dependent phosphorylation is consistent with the ion transport disturbances documented in cystic fibrosis tissues (for review, see(20)). These disturbances in ions change the concentration of molecules in the fluid within the ducts or organs (117).
4. CFTR Protein Structure
The CFTR protein is comprised of 1480 amino acids organized into 5 functional domains (87, 114). As other ABC transporters, CFTR has two membrane-spanning domains (TMD1 and TMD2), two nucleotide-binding domains (NBD1 and NBD2) and one regulatory domain (R) (Figure 1). For more insights regarding the structure of CFTR see the review by Patrick and Thomas (74). The two TMDs, each composed of 6 transmembrane segments, form the CFTR channel pore, and the two NBDs interact with nucleotides to regulate channel activity opening and closing of the TMDs (Figure 1) (74, 114). The R domain, through interactions with the N-terminal cytosolic region of TMD1, also controls the channel activity (12, 64, 114). NBDs are responsible for the binding and hydrolysis of the ATP, which causes a conformational change in the TMDs leading to the transport of substrates across cell membranes (95). CFTR mutations can occur in any of the five protein domains. However, many mutations occur in NBD1, including the ΔF508 mutation. The location of the CFTR mutations can affect the formation or function of the CFTR protein (Figure 2, Table 3) (114). In Table 3 we summarized the role of not only the major domains described for CFTR but also the connecting sequences which include: N-terminal, intracellular loops (ICL), extracellular loops (ECLs), transmembrane helixes 1 through 12, C-terminal domain. The N-terminal domain was shown to be involved in the folding and the trafficking of the CFTR protein through protein-protein interactions (e.g. syntaxin 1A) and mutations in this domain reduced the function of the channel (28, 65). Some transmembrane (TM) helices have been studied more than the others. For instance, the results from many studies suggested that transmembrane segments,TM1, TM2, TM6, TM11 and TM12 form the pore lining and regulate the pore function by selecting the anions (111, 112, 120), whereas TM5 plays a role in the anion binding (112). Also, V232D mutation in TM4 leads to a loss of function because it forms a bond with Q207 in TM3 which does not occur in the WT form of CFTR (73, 103). The mutation V232D is not the only mutation that involves a change from a neutral/hydrophobic residue to a polar or charged residue, causing CF. Therien et al, studied more than 31 mutations in TMD1 including all 6 TM helices and concluded that CFTR mutations in the TMs lead to a loss of function through the formation of membrane-buried interhelical hydrogen bonds (103). Extracellular Loops (ECLs) 1 through 6 represent about 4% of the CFTR protein, whereas 77% is in the cytoplasm and the rest, 19%, includes TM1 to 12. CF-associated mutations in the ECLs have been shown to affect channel gating (34), and interactions with extracellular anions (54, 121). For example, mutations D110H/E and R117C/H/L/P in ECL1 are associated with CF disease. These mutations affect the stability of the CFTR ion pore, resulting in reduced conductance of CFTR (34, 42). Q890X and K892C mutations in ECL4 have been reported to affect channel gating and extracellular anion interaction (10, 29, 42). ECL4 is the only extracellular loop that contains N-linked glycosylation sites (N894 and N900) (10). Many studies have been performed on the function of CFTR’s intracytoplasmic loops (ICLs) including their roles in regulating inter-molecular interactions as well as CFTR interactions with other proteins. E193K and I148T mutations in ICL1 have been reported to affect the pore opening (11, 18). Recent studies showed that ICL 2/3-NBD2 interface and ICL1/4-NBD1 interface have a role in protein folding and processing in the ER (7, 56). In addition, S945L, H949Y and G970R mutations in ICL3, L1065P, R1070Q and Q1071P mutations in ICL4, and D1152H, D1154G and W1204X mutations in ICL5 have been shown to affect the conductance of CFTR (29, 94, 107),. As for the rest of the domains, ATP-binding events occurring in both NBDs allow the hydrolysis of intracellular ATP to ADP (6). This event allows the conformational changes, and that change in structure allows the CFTR channel to transition from an opened to closed state, thus controlling the gating kinetics of the channel (6). NBD1 was studied more than NBD2 due to the presence of frequent ΔF508 deletion of NBD1 in the CF patients. The ΔF508 is present in more than 70% of the CF patients, resulting in destabilization of the CFTR protein (6, 90, 119). Also, mutations ΔF508 and G551D modify the interactions between NBDs and NBDs–ICLs (6). Other well-studied NBD1 mutations such as G542X and G551D result in channel-gating problems (6, 90, 109, 119). Similarly, NBD2 mutations like N1303K, p.Ile1234_Arg1239del, G1244E, S1251N, S1255P, and G1349D are CF disease related, and are considered to be gating mutations (109). The R domain, along with NBDs, control the channel activity of CFTR. The activation of the channel is dependent on the phosphorylation by cyclic AMP-dependent protein kinase (PKA) (93). To date, more than 15 phosphorylation sites in the R domain have been attributed to PKA phosphorylation, which contribute in varying proportions to the response to activation of the CFTR channel (93). It has been reported that CFTR also can be phosphorylated by several other protein kinases including protein kinase C (PKC), casein kinase II, cyclic GMP activated protein kinase, and Src kinase (93). The R domain has multiple phosphorylation sites for PKC, which modulate PKA-induced domain-domain interactions (92). Many CF-related mutations like D648V, E664X, E656X and 2108delA in the R domain disrupt the normal function of the R domain, e.g. the transport of HCO3- in secretory epithelia and in CF (3, 14). The same authors showed that mutants reported to be associated with CF with pancreatic insufficiency do not support HCO3- transport, and those associated with pancreatic sufficiency show reduced HCO3- transport (14).

Figure 1.Activation of the CFTR channel. The cystic fibrosis transmembrane conductance regulator (CFTR) protein channel is a member of the ABC transporter superfamily. It acts in apical part of the epithelial cells as a plasma-membrane, cyclic AMP-activated chloride and bicarbonate anion. CFTR has two membrane-spanning domains (TMD1 and TMD2), two nucleotide-binding domains (NBD1 channel and NBD2) and one regulatory domain (R). CFTR is a key regulator for cell surface water-salt homeostasis of the apical membranes of epithelial cells in multiple organs including the pancreas that produces alkaline fluid in pancreatic ducts. The open status of the CFTR is initiated by ATP binding at the NBD domains. The activation of the channel is dependent on phosphorylation by cyclic AMP-dependent protein kinase (PKA) at multiple sites in the R domain. The magnitude of response to PKA is amplified by phosphorylation of CFTR by protein kinase C (PKC).

Figure 2. Classes of CFTR mutations. Class I mutations lead to no protein synthesis, which includes mutations that includes premature stop codons and nonsense mutations. Class II mutation include the most frequent mutation of CF disease, ΔF508, which lead to trafficking, improper folding, and processing defects of the CFTR protein. This class is the primary target in the CF research and the main target by the pharmaceuticals companies. Class III mutations affect the ATP binding at one of the 2 binding sites in the NBDs. The CFTR protein reaches the cell surface but the mutations render the CFTR channel nonfunctional which impairs the opening of the channel. Class IV mutations also involve CFTR protein reach the cell surface but with reduced ion passage through the channel because of the structural defect caused by the mutations in the CFTR channel. Class V mutations affect the amount of CFTR protein that reaches the cell surface because of a splicing problems or inefficient trafficking. Class VI mutations lead to a rapid turnover of the CFTR channel at the cell surface. Examples of CFTR mutations regarding their pancreatic defects, PI or PS.
The mutations mentioned in the table are representative of each class, but we have to keep in mind, according to the new classification some mutations are classified in more than one class which will result of having more than one defect.
Table 3. Domains structure of CFTR. A detailed description of the CFTR domains consist of a n-terminal, 6 intracellular loops (ICL), 6 extracellular loops (ECL), 12 transmembrane (TM) arranged into TMD1 and TMD2 consist of 6 TM for each domain, NBD1 and NBD2, and the c-terminal domain. The specified aa for each domain, their known function, positive charge residues, some mutations causing CF, and their effects on the pancreas are as well described.
PI: pancreas insufficient, PS: pancreas sufficient.
|
Domains |
Amino acids |
Functions |
Positive charge residues |
Mutations causing CF |
References |
Pancreas |
|
N-terminal |
1 to 81 |
Folding and/or trafficking Protein Interaction |
(28, 65) |
PI |
||
|
TM1 |
82 to 103 |
Regulation of pore function; Pore lining |
K95 |
(39, 111, 112, 120) |
PS/PI |
|
|
ECL1 |
104 to 117 |
Stability of the CFTR ion pore |
R117C/H/L/P |
(34, 42, 49, 111) |
PS/PI |
|
|
TM2 |
118 to 138 |
CFTR pore lining |
R134 |
(52, 111, 112, 120) |
PS/PI |
|
|
ICL1 |
139 to 194 |
Pore opening |
E193K, I148T |
(11, 18, 56) |
PS/PI |
|
|
TM3 |
195 to 215 |
ND |
Q207 form a bond with the mutation V232D |
(103) |
||
|
ECL2 |
216 to 220 |
(34) |
||||
|
TM4 |
221 to 241 |
Loss of function of pore |
V232D |
(26, 73, 103) |
PS/PI |
|
|
ICL2 |
242 to 307 |
Protein folding, Processing in the ER |
(7, 56) |
|||
|
TM5 |
308 to 328 |
Anion binding |
(112) |
|||
|
ECL3 |
329 |
ND |
||||
|
TM6 |
330 to 350 |
Pore lining; anion selectivity |
R334, K335, R347 |
(112, 120) |
PS/PI |
|
|
ICl2.5 |
351 to 432 |
ND |
||||
|
NBD1 |
433 to 586 |
Hydrolyzation of ATP, Channel opening, regulation of the sodium ion channel |
Delta(F508); G551D; G542X |
(90, 119) |
PI |
|
|
R |
587 to 859 |
Phosphorylation sites for PKA/PKC |
D648V, E664X, E656X and 2108delA |
(3) |
PI |
|
|
TM7 |
860 to 870 |
ND |
||||
|
ECL4 |
881 to 911 |
Glycosylation at N894 and N900 |
Q890X, K892C |
(10, 29, 42) |
||
|
TM8 |
912 to 932 |
ND |
S912L |
(29) |
||
|
ICL3 |
933 to 990 |
Conductance |
S945L, H949Y, G970R |
(29, 94) |
PS/PI |
|
|
TM9 |
991 to 1011 |
ND |
||||
|
ECL5 |
1012 |
ND |
||||
|
TM10 |
1013 to 1034 |
Processing |
R1030 |
PI |
||
|
ICl4 |
1035 to 1102 |
L1065P, R1070Q, Q1071P |
(29) |
|||
|
TM11 |
1103 to 1123 |
Pore lining; anion selectivity |
(39, 111, 112, 120) |
PS/PI |
||
|
ECL6 |
1124 to 1128 |
ND |
||||
|
TM12 |
1129 to 1150 |
Pore lining; anion selectivity |
M1137V, M1137R, I11139V and deltaM1140 |
(107, 112, 120) |
PS/PI |
|
|
ICL5 |
1151 to 1218 |
Conductance |
D1152H, D1154G, W1204X |
(29, 107) |
PS/PI |
|
|
NBD2 |
1219 to 1386 |
Maturation, gating |
N1303K, G1349D, G1244E, S1251N, S1255P, and G1349D |
(107) |
PI |
|
|
C-terminal |
1387 to 1480 |
ND |
5. Classification of CF Patients
Traditional classification
As mentioned above, more than 2023 CF mutations have been identified (http://www.genletsickkids.on.ca/cftr/) in the CFTR gene. These mutations are categorized based on the dysfunctions of CFTR at different levels of the maturation and function of the CFTR channels (Figure 2).Traditionally, these dysfunctions were divided into 6 groups based on function (Classes III, IV, and VI) and processing (Classes I, II, and V) of the CFTR (Figure 2) (51, 58, 108). The class I mutations includes a nonsense, frame-shift, or splicing mutations which prevent CFTR biosynthesis by introducing a premature termination codon (PTC). The most common mutation in this class is the G542X (Figure 2).
The class II mutations include a missense mutation which causes misfolding of the CFTR to lead to its degradation in the ER by quality-control machinery, resulting in the absence of functional protein at the cell surface. The most important mutation in this class is the ΔF508 presents in more than 90% of CF patients (Figure 2).
The class III mutations include a missense mutation which lead to a non-functioning CFTR at the cell surface resulting in unstable and reduced channel gating characterized by a lower open probability. The most common mutation representing this class is the G551D (Figure 2).
The class IV mutations include missense mutation which leads to a reduced CFTR channel conductance. The decrease in conductance is caused by an abnormal the conformation of the pore resulting in disruption of the ion flow. The most common mutation in this class is the R117H (Figure 2).
The class V mutations introduce splicing or promoter defects in the CFTR gene, resulting in a reduced amount of CFTR protein at the cell membrane caused by reduced protein synthesis. Those mutations affect the gene expression, but do not change the conformation of the channel. The most representative mutations in this class are the 3849+10kbC-T and A455E (Figure 2).
The class VI mutations include missense mutation which lead to a decrease in the CFTR stability. These mutations result in an accelerated turnover of CFTR protein at the cell membrane and reduced apical cell surface expression (108). The most representative mutation in this class is Q1412X (Figure 2).
Though more than 2023 mutations/variants have reported for CFTR, whether each of these can cause channel dysfunction and disease is largely unknown. However, studies to predict the functional consequences and clinical outcome of individual patients carrying these mutations are being conducted (24, 97). These interpretations of such studies have been challenged by the general lack of correlation between the genotype and the clinical severity (24) (Table 1).
New classification
The lack of correlation between the genotype and the phenotype of the CF patients led to a new classification based on the severity and the clinical symptoms of the CF patients. Recently, Dupuis et al, studied meconium ileus (MI) in CF patients, and reported that only a subset of patients with CF develop MI (24). Furthermore, MI demonstrates notable heritability. Although studies have shown that non-CFTR genes contribute to susceptibility, the CFTR genotype itself affects the occurrence of this complication; only patients with the more severe CFTR variants are at risk for MI (24). It was hypothesized that the susceptibility to MI is influenced by specific CFTR genotypes, and that the prevalence of MI can be used to discriminate among severe CFTR mutations (24). The pleiotropic molecular defects of a single mutation in the CFTR has limited the drug therapy effects for some mutants which have been categorized as class I, II, or II/IV (108). The authors proposed a modification of the traditional class I–VI CF mutations classification. This expanded classification of the major mechanistic categories (87, 114, 122) accommodates the unusually complex, combinatorial molecular/cellular phenotypes of CF alleles. The new classification consists of 31 possible classes of mutations, including the original classes I, II, III/IV, V, and VI, as well as their 26 combinations (108). For example, according to the expanded classification, G551D will be designated as a class III mutation as before (114), while ΔF508 will be classified as class II–III–VI, W1282X as class I–II–III–VI, P67L as class II–III, Q1412X as class III–VI and R117H as class II–III/IV, reflecting the composite defects in mutant CFTR biology (108). More evidence supporting the new classification came from a study by Vertex Pharmaceuticals where they tested 54 missense mutations and found that 24 of them have both processing and gating defects (106).
6. CFTR Function and Its Role in Pancreas
Cystic fibrosis and exocrine pancreas
As mentioned above, CFTR is predominantly expressed on the apical membrane of epithelial cells in the small pancreatic ducts. CFTR acts as a selective ion channel involved in chloride, bicarbonate (HCO3-), water transport across the apical membranes of epithelial cells in multiple organs including the pancreas that produces alkaline fluid in pancreatic ducts (30, 70, 117). HCO3- is an important ion in the pancreatic juice. It also facilitates solubilization of the digestive enzymes and mucins (52). Indeed, aberrant HCO3- transport has a crucial role in human diseases (79, 80). In his review, Quinton proposed that in CF patients, the HCO3- is required to form normal mucus. His explanation is that once granule is released, HCO3- sequesters Ca2+ and H+ ions away from the mucin anions to form a complex with them. Therefore, lack of secreted HCO3- in CF patients impairs Ca2+ removal, prevents normal mucin expansion, and promotes stasis of mucus in the ducts or on the luminal surfaces of affected organs (79). In addition, reduced secretion of HCO3- and chloride (Cl-) leads to a more acidic and viscous luminal content (39). MUC6 is one of the pancreatic mucins expressed by 13 weeks of gestation and shows a very similar distribution to that of CFTR. In addition, MUC6 mucin is the main constituent of the complexes that form in small ducts and cause obstruction (81). Therefore, CF patients carrying mutations in the CFTR gene, showed a lower pH, low flow of secretions and high protein concentration in the pancreas duct secretions, which lead to precipitates in the duct lumina that obstruction and injury (30, 117). Meyerholz and his colleagues showed in the CF pig model that the changes (obstruction) could be detected in gestations as early as week 17 (59). They showed that the site of obstruction ranged from the distal jejunum to the proximal spiral colon, similar to that reported in humans with meconium ileus (59, 63). The obstruction in the acini and ducts lead to dilatation which causes epithelial injury and destruction, inflammation, fibrosis and fatty infiltration (30, 41, 59). Tucker and colleagues reported that acinar plugs developed before mucous metaplasia and found that early acinar plugs are composed of zymogen granules and were distinct from mucus in pancreatic tissue of cystic fibrosis patients (104). These findings then indicate that zymogen material from the acinar cell, not mucus, may become inspissated in the acinus in early cystic fibrosis, and that subsequent mucous metaplasia occurs as the obstruction and exocrine atrophy progress (104).
CFTR and Hyperinflammation
As mentioned above airways of the lungs become colonized with bacteria and repeated pulmonary infections ensue. The recurrent infections and inflammation result in submucosal gland hypertrophy and excessive mucus secretion. The impaired mucociliary clearance and plugging of small airways cause progressive bronchiectasis and ultimately lead to respiratory failure (100). Many studies have been done to explain the cause of the hyperinflammation in the CF patients. Many have suggested that the balance between Th1, Th2, and Th 17 could play a role in the CF disease (31). It has been established that Th17 is known to be a key player in autoimmune diseases (40). In addition, the authors showed that Th17 is regulated by miR-183C via inhibition of Foxo1(40). For a long time, it has been thought that CFTR mutations in epithelia cells have an indirect effect on the immune system which causes the inflammation in the lungs, because of the colonization of bacteria in the lungs like P. aeruginosa, Burkholderia cenocepacia, and Mycobacterium abscessus which causes the infections in the CF patients (31). Recently, emerging evidence points the problem to be directly affecting the immune cells. Since it has been shown that CFTR is expressed in lymphocyte T cells (like Th2, Th17, and Tregs) and macrophages, so CFTR mutations causing CF may have a direct impact on these cells. Grumlli et al summarized in his review, that CFTR mutations have a direct effect on the T cells function which results in an enhanced Th2 response, a reduced Treg population and elevated Th17 response which translate by an increase of neutrophils and recruitments by IL-17 to the lung which leads to the destruction of alveoli in the lungs of CF patients (31). In addition to the lymphocytes and neutrophils, macrophages also has been shown to participate to the lung decay found in CF patients through activation of MMp12 (31). Understanding better the mechanisms behind the infections and the immune response could lead to a better drug therapy targeted to each patient depending on the severity of the CF disease.
Cystic fibrosis and endocrine pancreas
CF is also recognized to affect the endocrine pancreas. There is a correlation between glucose abnormalities, morbidity and mortality in CF patients (67, 69). Glucose abnormalities include cystic fibrosis-related diabetes (CFRD) and impaired glucose tolerance (IGT). CFRD is one complication in the CF patients occurring in more than 40 % of adults and 25% of adolescents, which is preceded by episodes of impaired glucose tolerance (19, 61, 67, 91). Both reduced insulin secretion and insulin resistance are observed in CFRD (36, 62, 66, 78). CFRD has characteristics of both type I and II diabetes and does not belong to either one of the diabetes classes. It is characterized by the loss of functional β cell mass and varying degrees of insulin resistance (Figure 3) (30, 62, 66). Mutations in CFTR that lead to both a decrease in islet cell mass and dysfunction in the β cell are the cause of CFRD (17, 67, 86). It is believed that cross-talk between pancreatic exocrine and endocrine components can also contribute to the CFDR (Figure 3). Destruction of pancreatic exocrine tissue caused by a decrease in the islet cell mass evolves to dysfunctional endocrine β cells (5). β cell dysfunction may be caused by increased oxidative and endoplasmic reticulum (ER) stress which are associated with CFRD (27, 77, 84, 118). It is very well documented that glucose deprivation leads to ER stress (118). Some CFTR mutations (Like ΔF508) can cause the accumulation of unfolded proteins in the ER, triggering an evolutionarily conserved response, termed the unfolded protein response (UPR) (2). In addition, aberrant Ca2+ regulation in the ER lumen causes protein unfolding and rapid degradation of mutated CFTR proteins may contributes to the complex multi-organ CF pathology (48). It is known that ER stress triggers β cell death typically by apoptosis when protein misfolding is persistent or excessive (76). As Harding at al stated, the special sensitivity of insulin-producing cells to a mutation (like CFTR mutations) that affects a signaling protein responsive to ER stress may also be relevant to the development of more common forms of human diabetes mellitus. The major abnormality in most patients with CFRD is resistance to the action of insulin. However, glucose intolerance develops only after β cell decompensation renders the endocrine pancreas unable to keep up with the demand imposed by IR (37). Because of their high rates of protein synthesis, β-cells are particularly susceptible to ER stress, which may trigger CFRD (67). The expression of CFTR was reported to be required in β-cells for glucose-induced secretion. Therefore, CFTR plays a significant role in the normal function of pancreatic β-cells (85). Ntimbane and his colleagues have summarized the factors leading to an abnormal glucose homeostasis in CF patients: (a) impairment of β-cell function with progressive fibrosis of islets of Langerhans with resultant distortion, ischaemia, cell death and a decrease in islet numbers; (b) impairment of other islet cell functions; (c) impairment of the insulinotropic gut hormone secretin; (d) changes in insulin sensitivity; and (e) altered insulin clearance rate (67). The exact cause and mechanism of CFRD are still largely unknown. The most probable cause of CFRD is a combination of many events and unlikely to be attributable to one defect. The clinical effects and disease states associated with CF patients with CFRD include: chronic pancreatic inflammation, dysfunction of the immune system, oxidative stress, impaired insulin production and secretion, variable state of IR and altered entero–insular axis hormones (30, 67).

Figure 3. CF and pancreas defects. Schematic representation of the cross talk between exocrine and endocrine of the pancreas. Defects in the exocrine pancreas leads to PI and PS, causing the anions imbalance. Defects in the endocrine pancreas leads to CFRD. As summarized in this diagram, many factors, defects and the crosstalk between endocrine and exocrine leads to the phenotypes described in the CF patients including PI, PS, and CFRD.
Insulin secretion and CFTR
All the proposed causes mentioned above are supported by evidence to explain the pathogenesis of impaired insulin secretion in CFRD. An additional cause not mentioned above is the expression and direct effect of CFTR on insulin secretion in the β-cells (26, 32, 47, 102). The expression of CFTR has been reported in cultured β-cells derived from mice (MIN6) or rat (RINm5F) (26, 32, 68). But the earliest report came in 2007 from studies by Boom and his colleagues showing the expression of CFTR protein in rat islet cells and the significantly higher level in non-beta than in beta- cell populations (8). They also showed by immunohistochemistry studies that CFTR expression also occurs in glucagon-secreting alpha-cells (8). Guo and his colleagues demonstrated that glucose-induced whole-cell currents, membrane depolarization, electrical bursts or action potentials, Ca2+ oscillations and insulin secretion in β-cells are dependent on CFTR, indicating an essential role of CFTR in the regulation of insulin secretion (32). Their studies showed that specific inhibitors of CFTR (GlyH-101 and CFTRinh-172) blocked a CFTR Cl- gating needed for insulin secretion in primary β-cells and ΔF508-CFTR mutant mouse islets (32). In addition, Edlund and her colleagues detected small CFTR conductance in both human and mouse beta-cells. The augmentation of insulin secretion by activation of CFTR by cAMP (forskolin or GLP-1) in the presence of glucose was significantly inhibited by the specific CFTR inhibitors. They also demonstrated reduced cAMP-dependent exocytosis upon CFTR-inhibition, concomitant with fewer docked insulin granules (26). These reports and others from the patients with CFTR mutations showed insufficiency of secreted insulin. However, these studies did not describe the molecular mechanism that cause a decrease in insulin secretion. In addition, to the role of CFTR in regulating insulin secretion and exocytosis after glucose-induced membrane depolarization leading to insulin secretion, the study also demonstrated that CFTR molecules act upstream of the chloride channel Anoctamin 1 (ANO1; TMEM16A) in the regulation of cAMP- and glucose-stimulated insulin secretion (26). Thus the impaired insulin secretion seen in patients with CF would be caused by the lack of glucose-induced Cl- efflux through both CFTR Cl- channels and ANO1 due to a decreased membrane depolarization (26, 32). In summary these study showed that CFTR is an important regulator of pancreatic β-cell insulin secretion, exocytosis, and membrane depolarization, and is induced by glucose via elevation of cytosolic Ca2+ concentration (32, 47, 58).
Marunaka in his recent review summarized new studies connecting the role of CFTR and ANO1 in insulin secretion (58). Briefly, it has been established that intracellular Cl- concentration ([Cl−]) is a very useful marker of channel activity. In these recent studies, the authors showed that [Cl−] measured using N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide (MQAE) is about 100 mM under the basal condition in RINm5F β cell line, and application of CFTRinh-172 (an inhibitor of the CFTR channel) increased [Cl−] about 26 mM (32). This means that CFTR indeed mediates Cl- efflux under basal condition, which may maintain a relatively depolarized membrane potential in the β-cells at rest, and the electrochemical potential of Cl− in the intracellular space is larger than that in the extracellular space (32, 58). This was confirmed by showing that membrane potentials of pancreatic β cells expressing wild-type CFTR Cl− channels are −61~−67 mV [70], but hyperpolarized to −75 mV when using CFTRinh-172 or ΔF508 (32). Thus, CFTR has an important role in determining the resting membrane potential of the β-cells by acting as a Cl− channel to maintain the membrane depolarization (32). The same study also showed that ΔF508 CFTR Cl− channel decreases membrane depolarization induced by glucose and increases [Ca2+] due to the activation of voltage-dependent Ca2+ channels (32, 58). In his review, Marunaka concludes that the loss of CFTR function leads to insulin insufficiency which is caused by the higher intracellular Cl− electrochemical potential in pancreatic β cells. “In general, Cl− uptake into the intracellular space is mediated via active Cl− transporting systems, such as Na+-Cl− cotransporter (NCC) and/or NKCC, driven by the Na+,K+-ATPase-generated Na+ chemical potential difference between the intracellular and extracellular spaces: the intracellular Na+ chemical potential < the extracellular Na+ chemical potential. Therefore, if we could increase the [Cl−] by elevating the NCC- and/or NKCC-mediated Cl− uptake, the insufficiency of insulin secretion would be improved via membrane depolarization due to an elevation of Cl− efflux from pancreatic β cells of ΔF508 CFTR-expressing CF patients.
Recent studies contradict the findings by Guo et al, by presenting some evidence using the ferret’s pancreas that β-cells do not express CFTR (102). The authors showed that CFTR RNA is expressed in exocrine and not in endocrine cell types of islets and pancreas. They used a different approach, smFISH, to show the expression of CFTR. WT and CFTR-KO neonatal ferret pancreas were used to perform CFTR and INS dual smFISH. As expected, the INS was present in both genotypes, but CFTR was present in the WT pancreas (102). More importantly, CFTR RNA was not co-expressed in INS (β-cell), GCG (α-cell), PPY (PP cell), or SST (δ-cell) expressing cells but was expressed in KRT7-expressing ductal cells in the WT pancreas. Similarly, same findings were shown in dissociated cells from isolated adult ferret and human islets (102). These findings contradict the previous findings by Guo et al, and demonstrate that exocrine-derived duct cells associated with isolated islets express the highest levels of CFTR and support a mechanism by which CFTR dependent duct/islet crosstalk might influence β-cell insulin secretion (102).
Although there is evidence that supported each of these contradicting studies, a clear resolution to the question of whether CFTR directly or indirectly functions within the β-cells or other islet cell types to support insulin secretion needs further clarifications.
Role of glucose transporters (GLUTs) in CF and their impact in the pancreas
As mentioned above, glucose abnormalities in CF include CFRD and impaired glucose tolerance. The relationship between CFTR and the causes of CFRD is still not very well established. Studies are also emerging regarding the implication of GLUT transporters in developing the diabetic state in CF patients (53). In studies regarding obesity and diabetes, it was reported that CFTR was significantly decreased, while GLUT5 and Villin were increased in the jejunum (53). It is known that CFTR Cl– channel provide the major route for Cl– exit across the apical membrane in normal murine intestine and disturbance in the anion exchange and recycling of K+ is thought to be one of the causes of diabetes (53).
Recently it was reported that GLUT4 and apical insulin are expressed in normal human airway epithelial cells (60).The authors also showed that cells expressing F508del-CFTR have impaired glucose uptake, elevating action on the trans epithelial resistance, and diminishing action on paracellular flux of small molecules after insulin stimulation (60). In a different study, GLUT4 subfractionation demonstrated that, despite insulin stimulation, the GLUT-4 content of intracellular-associated subfraction was significantly higher in CF subjects compared with controls, corresponding to significantly lower GLUT-4 content in cell surface-associated subfraction (35). These findings are consistent with the abnormal subcellular localization of GLUT-4. Impairment of GLUT-4 translocation in CF correlated with higher TNF-α levels in all CF subjects than in controls (35). CF patients that have CFRD with a decreased insulin secretion could be explained by the elevation of TNF-α and impaired translocation of GLUT-4. In addition, the results indicate that the function of CFTR Cl− channels is required for insulin to stimulate glucose uptake, elevate the transepithelial resistance, and diminish the paracellular flux of small molecules in airway epithelial cells (58).
7. Pancreatic Defect and CFTR
In general, the exocrine pancreatic disease and its progression correlates well with the genetic factors of the CF patients (1, 49). CF patients have been divided into two classes, pancreatic insufficient (PI) and pancreatic sufficient (PS) (1, 46, 49). Approximately 85% of patients with CF have PI which is categorized as the “severe” CF phenotype, the rest of 15% are PS patients and thus “mild” CF phenotype. The exocrine pancreas in PI patients no longer secretes the required digestive enzymes. Therefore, CF patients often require an oral pancreatic enzyme supplement each meal (117). Pancreatic damage from CF can be detected in utero in subjects with PI and show obstruction of the small ducts and acini which will lead to the destruction of the pancreas with only a few islets or ducts left in a sea of adipose tissue (30). In contrast, PS-CF patients do have pancreatic damage, as measured by the high levels of serum immunoreactive trypsinogen (IRT), but retain normal digestion due to a sufficient endogenous function of exocrine pancreatic ducts (30, 117). As we have mentioned above, CF patients are classified into six traditional classes from I to VI based on their CFTR mutations. The exocrine pancreatic phenotypes PI and PS are directly linked to genotype (1, 46, 49, 114).
Wilschanski and his colleagues describe that CF patients homozygous or compound heterozygous for severe alleles belonging to classes I, II, III, or VI confer PI. Whereas a mild class IV or V allele sustains pancreatic function in a dominant fashion even if the second mutation is severe and falls into PS (117). They go further and explained that this observation appears plausible because all known mild alleles belong to class IV or V, all of which are (or predicted to be) associated with some residual chloride channel activity at the epithelial apical membranes (117). On the other hand, in a recent review by Gibson-Corley and his colleagues, they described the PI CF patients to be in the classes I, II, III, IV and VI because they have mutations that render CFTR to be absent or non-functional (30). The remainder of the patients belonging to class V or mild class IV considered PS, due to less severe CFTR mutations (30).
In both reviews, it was mentioned that this classification of the PI and PS do not entirely fit into the six classes of CFTR mutations. Those who are considered PS still show pancreatic destruction as the serum level of IRT is elevated but will not require enzyme replacement for normal digestion (30). Some class I mutations with the stop codon at the end of the gene are CF patients with PS (117). In addition, a small portion (~3%) of CF patients with a severe mutation on both alleles are considered PS at diagnosis, but eventually transition from PS to PI (25, 117). Another example, G85E a missense mutation and few other mutations do show variable pancreatic phenotypes (117). PS CF patients are more susceptible to developing pancreatitis than the PI patients (25). It is known that pancreatic ducts have an essential part in CF and chronic pancreatitis, and only PS patients develop pancreatitis, suggesting that partially impaired the function of pancreatic ducts is retained in PS patients (38). Druno and colleagues showed that there is a strong correlation between genotype and phenotype in patients with CF and pancreatitis (25). They showed from a CF cohort study of about 1000 patients followed over a period of 30 years that PS CF patients carry at least one mild mutant allele and are at a significant risk of developing pancreatitis. Symptoms of pancreatitis may precede the diagnosis of CF. Pancreatitis is associated with an otherwise mild CF phenotype (25). A larger CF cohort study of about 10071 patients had reported that out of 331 patients with PS, 34 cases had pancreatitis, where the occurrence of pancreatitis among patients with PI was 15 cases out of 2971 patients (21). More evidence goes toward the correlation of PS CF patients and the development of pancreatitis, with a novel pancreatic insufficiency prevalence score where they divided the patients into 3 groups: severe, moderate-severe, and mild, with the mild mutations more susceptible to the risk of developing pancreatitis (71, 117). The new classification of CFTR mutations into 31 new classes reflects the composite defects in mutant CFTR biology (108). We will need to take into considerations the complexity of the disease and the severity of the CFTR genotype and their relationship with risk of pancreatitis. In a very recent study, for example, three sibling patients with a novel missense mutation, the R248G in exon 6 of the CFTR gene, present a recurrent acute pancreatitis (110). A similar missense mutation, R248T, has been previously reported as a mild CFTR-RD mutation that is not associated with pancreatitis. The R248G mutation may alter the normal function of CFTR more than the R248T mutation based on the clinical phenotypes of the three patients (110). As the authors conclude, future structure-functional studies on the CFTR protein can provide further insight into the impact of the R248G mutation at the molecular level.
8. CFTR Mutations and Pancreatitis
It is very well established by now that CFTR is a key protein in the pancreatic duct, which regulates the exchange of anions between the luminal surface and the cytoplasm of the duct cells. As mentioned, in CF patients the pancreas is one of the first organs to fail because mutations in CFTR play a critical role in pancreatic pathophysiology. The large number of mutations known to date in CFTR lead scientists to tackle this complex disease with so many symptoms from a different angle, i.e.,to make a specific correlation between CFTR mutations with certain symptoms such as pancreatitis in its both forms chronic and acute to determine the severity of the disease. In an Austrian cohort study of 133 pancreatitis patients, the frequency of CFTR mutations was 11.2% (123). In patients classified as ‘idiopathic definitive chronic pancreatitis,’ the frequency of mutations was 12.7%, whereas patients with ‘acute pancreatitis’ or ‘possible chronic pancreatitis’, had a frequency of CFTR mutations of 10% and 9.1%, respectively (123). The authors concluded, the frequency of CFTR mutations is highest in patients with definitive chronic pancreatitis and may, therefore, be regarded as a risk factor for the development of chronic pancreatitis (123). Another large Canadian CF cohort study of 2481 subjects with PS-CF (with and without pancreatitis) showed some correlation between the severity of CFTR genotype and the risk of pancreatitis (71). They showed that patients carrying mild mutations are more likely to develop pancreatitis than those who had moderate -severe mutations (71). Therefore, patients with mild mutations had 71% increase in the risk of developing pancreatitis at any given time than those with moderate-severe mutations (71). Thus, approximately 20% of PS-CF patients develop pancreatitis (71). Coffey at al, in their review summarized the complexity of the correlation between the CFTR mutations and the development of pancreatitis by categorizing the mutations into four groups based on the clinical status of the patients: (i) CF-causing mutations, (ii) mutations associated with CFTR-related disease, (iii) mutations with no known clinical consequence, and (iv) mutations with unknown clinical relevance (15). Also, Ooi and colleagues found that certain diseases that resemble CF at an organ-specific level (e.g. pancreatitis) are also strongly associated with mutations in the CFTR gene (15, 72) In conclusion, pancreatitis in the CF patients and the relationship with multiple mutations of CFTR are very complex due to the multiple levels of the disease symptoms, which are different from mutation to another due to the extended classification of those different mutations into the 27 different classes according to the new classification.
9. Conclusion
In CF patients the pancreas is one of the first organs to fail because mutations in CFTR have a critical role in pancreatic pathophysiology. CFTR is the key regulator of the pancreatic duct that regulates the anion exchange between the luminal surface and the cytoplasm of the duct cells. The large number of CFTR mutations are leading scientists to approach an understanding of their functional impact from a different angle with the goal of making a specific correlation between CFTR mutations and certain symptoms such as PI and PS. The lack of correlation between the genotype and the phenotype of the CF patients has led to a new classification (31 possible classes of mutations) based on the severity and the clinical symptoms of the CF patients. The pleiotropic molecular defects of a single mutation in CFTR has limited the effects of drug therapy for some mutants which have been categorized as class I, II, or II/IV (108). The expanded classification of the major mechanistic categories will accommodate the unusually complex, combinatorial molecular/ cellular phenotypes of CF alleles. In addition to the new proposed classification, one more level of complexity is to categorize the mutations into four groups based on the clinical status of the patients: (i) CF-causing mutations, (ii) mutations associated with CFTR-related disease, (iii) mutations with no known clinical consequence, and (iv) mutations with unknown clinical relevance (15). All the recent discoveries and the new hypothesis will help shed light on the complex CF disease from a new perspective, which will help develop a new combined therapy to rectify the mutation or mutations at different levels of CFTR defects.
10. Acknowledgements
We thank Dr. XueQun Chen for assistance in reading, Jawad Bouhamdan and Noor Charara for assistance in English editing. This work was supported by grants to Fei Sun, from the National Heart, Lung, & Blood Institute:1RO1HL133162, and the cystic Fibrosis Foundation: SUN15XXO.
11. References
- Ahmed N, Corey M, Forstner G, Zielenski J, Tsui LC, Ellis L, et al. Molecular consequences of cystic fibrosis transmembrane regulator (CFTR) gene mutations in the exocrine pancreas. Gut 52(8): 1159-1164,2003. PMID: 12865275.
- Araki E, Oyadomari S and Mori M. Impact of endoplasmic reticulum stress pathway on pancreatic beta-cells and diabetes mellitus. Exp Biol Med (Maywood) 228(10): 1213-1217,2003. PMID: 14610263.
- Aznarez I, Chan EM, Zielenski J, Blencowe BJ and Tsui LC. Characterization of disease-associated mutations affecting an exonic splicing enhancer and two cryptic splice sites in exon 13 of the cystic fibrosis transmembrane conductance regulator gene. Hum Mol Genet 12(16): 2031-2040,2003. PMID: 12913074.
- Ballard S and Spadafora D. Fluid secretion by submucosal glands of the tracheobronchial airways. Respir Physiol Neurobiol. 159: 271-277,2008. PMID: 17707699.
- Barrio R. Management of endocrine disease: Cystic fibrosis-related diabetes: novel pathogenic insights opening new therapeutic avenues. Eur J Endocrinol 172(4): R131-141,2015. PMID: 25336504.
- Belmonte L and Moran O. On the interactions between nucleotide binding domains and membrane spanning domains in cystic fibrosis transmembrane regulator: A molecular dynamic study. Biochimie 111: 19-29,2015. PMID: 25640670.
- Billet A, Mornon JP, Jollivet M, Lehn P, Callebaut I and Becq F. CFTR: effect of ICL2 and ICL4 amino acids in close spatial proximity on the current properties of the channel. J Cyst Fibros 12(6): 737-745,2013. PMID: 23478129.
- Boom A, Lybaert P, Pollet JF, Jacobs P, Jijakli H, Golstein PE, et al. Expression and localization of cystic fibrosis transmembrane conductance regulator in the rat endocrine pancreas. Endocrine 32(2): 197-205,2007. PMID: 18040894.
- Brennan AL and Beynon J. Clinical Updates in Cystic Fibrosis–Related Diabetes. Semin Respir Crit Care Med 36(02): 236-250,2015. PMID: 25826591
- Broadbent SD, Wang W and Linsdell P. Interaction between 2 extracellular loops influences the activity of the cystic fibrosis transmembrane conductance regulator chloride channel. Biochem Cell Biol 92(5): 390-396,2014. PMID: 25253636.
- Caputo A, Hinzpeter A, Caci E, Pedemonte N, Arous N, Di Duca M, et al. Mutation-specific potency and efficacy of cystic fibrosis transmembrane conductance regulator chloride channel potentiators. J Pharmacol Exp Ther 330(3): 783-791,2009. PMID: 19491324.
- Chappe V, Irvine T, Liao J, Evagelidis A and Hanrahan JW. Phosphorylation of CFTR by PKA promotes binding of the regulatory domain. EMBO J 24(15): 2730,2005. PMID: 16001079
- Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, et al. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63(4): 827-834,1990. PMID: 1699669.
- Choi JY, Muallem D, Kiselyov K, Lee MG, Thomas PJ and Muallem S. Aberrant CFTR-dependent HCO3- transport in mutations associated with cystic fibrosis. Nature 410(6824): 94-97,2001. PMID: 11242048.
- Coffey MJ and Ooi CY. Pancreatitis in Cystic Fibrosis and CFTR-Related Disorder. Acute Pancreatitis. L Rodrigo. Rijeka, InTech: Ch. 06, 2012.
- Colledge WH, Abella BS, Southern KW, Ratcliff R, Jiang C, Cheng SH, et al. Generation and characterization of a delta F508 cystic fibrosis mouse model. Nat Genet 10(4): 445-452,1995. PMID: 7545494.
- Consortium CFG-P. Correlation between genotype and phenotype in patients with cystic fibrosis. N Engl J Med 329(18): 1308-1313,1993. PMID: 8166795.
- Cremonesi L, Ferrari M, Belloni E, Magnani C, Seia M, Ronchetto P, et al. Four new mutations of the CFTR gene (541delC, R347H, R352Q, E585X) detected by DGGE analysis in Italian CF patients, associated with different clinical phenotypes. Hum Mutat 1(4): 314-319,1992. PMID: 1284538.
- Cucinotta D, De Luca F, Scoglio R, Lombardo F, Sferlazzas C, Di Benedetto A, et al. Factors affecting diabetes mellitus onset in cystic fibrosis: evidence from a 10-year follow-up study. Acta Paediatr 88(4): 389-393,1999. PMID: 10342535.
- Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet 16(1): 45-56,2015. PMID: 25404111.
- De Boeck K, Weren M, Proesmans M and Kerem E. Pancreatitis Among Patients With Cystic Fibrosis: Correlation With Pancreatic Status and Genotype. Pediatrics 115(4): e463,2005. PMID: 15772171.
- Delaney SJ, Alton EW, Smith SN, Lunn DP, Farley R, Lovelock PK, et al. Cystic fibrosis mice carrying the missense mutation G551D replicate human genotype-phenotype correlations. EMBO J 15(5): 955-963,1996. PMID: 8605891.
- Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE and Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 358(6389): 761-764,1992. PMID: 1380673.
- Dupuis A, Keenan K, Ooi CY, Dorfman R, Sontag MK, Naehrlich L, et al. Prevalence of meconium ileus marks the severity of mutations of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene. Genet Med 18(4): 333-340,2016. PMID: 26087176.
- Durno C, Corey M, Zielenski J, Tullis E, Tsui L-C and Durie P. Genotype and phenotype correlations in patients with cystic fibrosis and pancreatitis. Gastroenterology 123(6): 1857-1864,2002. PMID: 12454843
- Edlund A, Esguerra JL, Wendt A, Flodstrom-Tullberg M and Eliasson L. CFTR and Anoctamin 1 (ANO1) contribute to cAMP amplified exocytosis and insulin secretion in human and murine pancreatic beta-cells. BMC Med 12: 87,2014. PMID: 24885604.
- Galli F, Battistoni A, Gambari R, Pompella A, Bragonzi A, Pilolli F, et al. Oxidative stress and antioxidant therapy in cystic fibrosis. Biochim Biophys Acta 1822(5): 690-713,2012. PMID: 22226887.
- Gene GG, Llobet A, Larriba S, de Semir D, Martinez I, Escalada A, et al. N-terminal CFTR missense variants severely affect the behavior of the CFTR chloride channel. Hum Mutat 29(5): 738-749,2008. PMID: 18306312.
- Ghanem N, Costes B, Girodon E, Martin J, Fanen P and Goossens M. Identification of eight mutations and three sequence variations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Genomics 21(2): 434-436,1994. PMID: 7522211.
- Gibson-Corley KN, Meyerholz DK and Engelhardt JF. Pancreatic pathophysiology in cystic fibrosis. J Pathol 238(2): 311-320,2016. PMID: 26365583.
- Grumelli S IG, Castro GR Consequences of cystic fibrosis transmembrane regulator mutations on inflammatory cells. Pulm Crit Care Med 1(2),2016. doi: 10.15761/PCCM.1000110
- Guo JH, Chen H, Ruan YC, Zhang XL, Zhang XH, Fok KL, et al. Glucose-induced electrical activities and insulin secretion in pancreatic islet beta-cells are modulated by CFTR. Nat Commun 5: 4420,2014. PMID: 25025956.
- Hameed S, Jaffe A and Verge CF. Advances in the detection and management of cystic fibrosis related diabetes. Curr Opin Pediatr 27(4): 525-533,2015. PMID: 26087430.
- Hammerle MM, Aleksandrov AA and Riordan JR. Disease-associated mutations in the extracytoplasmic loops of cystic fibrosis transmembrane conductance regulator do not impede biosynthetic processing but impair chloride channel stability. J Biol Chem 276(18): 14848-14854,2001. PMID: 11278813.
- Hardin DS, Leblanc A, Marshall G and Seilheimer DK. Mechanisms of insulin resistance in cystic fibrosis. Am J Physiol Endocrinol Metab 281(5): E1022-1028,2001. PMID: 11595659.
- Hardin DS and Moran A. Diabetes mellitus in cystic fibrosis. Endocrinol Metab Clin North Am 28(4): 787-800, ix,1999. PMID: 10609120.
- Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, et al. Diabetes Mellitus and Exocrine Pancreatic Dysfunction in Perk−/− Mice Reveals a Role for Translational Control in Secretory Cell Survival. Molecular Cell 7(6): 1153-1163,2001. PMID: 11430819.
- Hegyi P and Rakonczay Z, Jr. The role of pancreatic ducts in the pathogenesis of acute pancreatitis. Pancreatology 15(4 Suppl): S13-17,2015. PMID: 25921231.
- Hohwieler M, Perkhofer L, Liebau S, Seufferlein T, Muller M, Illing A, et al. Stem cell-derived organoids to model gastrointestinal facets of cystic fibrosis. United European Gastroenterol J 5(5): 609-624,2017. PMID: 28815024.
- Ichiyama K, Gonzalez-Martin A, Kim BS, Jin HY, Jin W, Xu W, et al. The MicroRNA-183-96-182 Cluster Promotes T Helper 17 Cell Pathogenicity by Negatively Regulating Transcription Factor Foxo1 Expression. Immunity 44(6): 1284-1298,2016. PMID: 27332731.
- Imrie JR, Fagan DG and Sturgess JM. Quantitative evaluation of the development of the exocrine pancreas in cystic fibrosis and control infants. Am J Path 95(3): 697-707,1979. PMID: 453330.
- Infield DT, Cui G, Kuang C and McCarty NA. Positioning of extracellular loop 1 affects pore gating of the cystic fibrosis transmembrane conductance regulator. Am J Physiol Lung Cell Mol Physiol 310(5): L403-414,2016. PMID: 26684250.
- Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL and Riordan JR. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 83(1): 129-135,1995. PMID: 7553864.
- Kamaruzaman NA, Kardia E, Kamaldin N, Latahir AZ and Yahaya BH. The rabbit as a model for studying lung disease and stem cell therapy. Biomed Res Int 2013: Art ID 691830,2013. PMID: 23653896.
- Keiser NW and Engelhardt JF. New animal models of cystic fibrosis: what are they teaching us? Curr Opin Pulm Med 17(6): 478-483,2011. PMID: 21857224.
- Kerem E, Corey M, Kerem BS, Rommens J, Markiewicz D, Levison H, et al. The relation between genotype and phenotype in cystic fibrosis--analysis of the most common mutation (delta F508). N Engl J Med 323(22): 1517-1522,1990. PMID: 2233932.
- Koivula FN, McClenaghan NH, Harper AG and Kelly C. Islet-intrinsic effects of CFTR mutation. Diabetologia 59(7): 1350-1355,2016. PMID: 27033560.
- Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 10(12): 524-530,2000. PMID: 11121744.
- Kristidis P, Bozon D, Corey M, Markiewicz D, Rommens J, Tsui LC, et al. Genetic determination of exocrine pancreatic function in cystic fibrosis. Am J Hum Genet 50(6): 1178-1184,1992. PMID: 1376016.
- Kunzelmann K, Schreiber R and Hadorn HB. Bicarbonate in cystic fibrosis. J Cyst Fibros 66: 653-662,2017. PMID: 28732801.
- Lavelle GM, White MM, Browne N, McElvaney NG and Reeves EP. Animal Models of Cystic Fibrosis Pathology: Phenotypic Parallels and Divergences. BioMed Res Int 2016: 5258727,2016. PMID: 27340662.
- Lee MG, Ohana E, Park HW, Yang D and Muallem S. Molecular mechanism of pancreatic and salivary gland fluid and HCO3 secretion. Physiol Rev 92(1): 39-74,2012. PMID: 22298651.
- Leung L, Kang J, Rayyan E, Bhakta A, Barrett B, Larsen D, et al. Decreased basal chloride secretion and altered cystic fibrosis transmembrane conductance regulatory protein, Villin, GLUT5 protein expression in jejunum from leptin-deficient mice. Diabetes Metab Syndr Obes 7: 321-330,2014. PMID: 25092993.
- Li MS, Cowley EA and Linsdell P. Pseudohalide anions reveal a novel extracellular site for potentiators to increase CFTR function. Br J Pharmacol 167(5): 1062-1075,2012. PMID: 22612315.
- Liu J, Walker NM, Ootani A, Strubberg AM and Clarke LL. Defective goblet cell exocytosis contributes to murine cystic fibrosis-associated intestinal disease. J Clin Invest 125(3): 1056-1068,2015. PMID: 25642775.
- Loo TW and Clarke DM. The Transmission Interfaces Contribute Asymmetrically to the Assembly and Activity of Human P-glycoprotein. J Biol Chem 290(27): 16954-16963,2015. PMID: 25987565.
- Mall M, Grubb BR, Harkema JR, O'Neal WK and Boucher RC. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med 10(5): 487-493,2004. PMID: 15077107.
- Marunaka Y. The Mechanistic Links between Insulin and Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Cl− Channel. Int J Mol Sci 18(8): E1767,2017. PMID: 28805732.
- Meyerholz DK, Stoltz DA, Pezzulo AA and Welsh MJ. Pathology of gastrointestinal organs in a porcine model of cystic fibrosis. Am J Pathol 176(3): 1377-1389,2010. PMID: 20110417.
- Molina SA, Moriarty HK, Infield DT, Imhoff BR, Vance RJ, Kim AH, et al. Insulin signaling via the PI3-kinase/Akt pathway regulates airway glucose uptake and barrier function in a CFTR-dependent manner. Am J Physiol Lung Cell Mol Physiol 312(5): L688-l702,2017. PMID: 28213469.
- Moran A, Doherty L, Wang X and Thomas W. Abnormal glucose metabolism in cystic fibrosis. J Pediatr 133(1): 10-17,1998. PMID: 9672504.
- Moran A, Hardin D, Rodman D, Allen HF, Beall RJ, Borowitz D, et al. Diagnosis, screening and management of cystic fibrosis related diabetes mellitus: a consensus conference report. Diabetes Res Clin Pract 45(1): 61-73,1999. PMID: 10499886.
- Mushtaq I, Wright VM, Drake DP, Mearns MB and Wood CB. Meconium ileus secondary to cystic fibrosis. The East London experience. Pediatr Surg Int 13(5-6): 365-369,1998. PMID: 9639619.
- Naren AP, Cormet-Boyaka E, Fu J, Villain M, Blalock JE, Quick MW, et al. CFTR Chloride Channel Regulation by an Interdomain Interaction. Science 286(5439): 544,1999. PMID: 10521352.
- Naren AP, Quick MW, Collawn JF, Nelson DJ and Kirk KL. Syntaxin 1A inhibits CFTR chloride channels by means of domain-specific protein-protein interactions. Proc Natl Acad Sci U S A 95(18): 10972-10977,1998. PMID: 9724814.
- Nathan BM, Laguna T and Moran A. Recent trends in cystic fibrosis-related diabetes. Curr Opin Endocrinol Diabetes Obes 17(4): 335-341,2010. PMID: 20489612.
- Ntimbane T, Comte B, Mailhot G, Berthiaume Y, Poitout V, Prentki M, et al. Cystic fibrosis-related diabetes: from CFTR dysfunction to oxidative stress. Clin Biochem Rev 30(4): 153-177,2009. PMID: 20011209.
- Ntimbane T, Mailhot G, Spahis S, Rabasa-Lhoret R, Kleme ML, Melloul D, et al. CFTR silencing in pancreatic beta-cells reveals a functional impact on glucose-stimulated insulin secretion and oxidative stress response. Am J Physiol Endocrinol Metab 310(3): E200-212,2016. PMID: 26625901.
- Ode KL and Moran A. New insights into cystic fibrosis-related diabetes in children. Lancet Diabetes Endocrinol 1(1): 52-58,2013. PMID: 24622267.
- Olivier AK, Gibson-Corley KN and Meyerholz DK. Animal models of gastrointestinal and liver diseases. Animal models of cystic fibrosis: gastrointestinal, pancreatic, and hepatobiliary disease and pathophysiology. Am J Physiol Gastrointest Liver Physiol 308(6): G459-471,2015. PMID: 25591863.
- Ooi CY, Dorfman R, Cipolli M, Gonska T, Castellani C, Keenan K, et al. Type of CFTR mutation determines risk of pancreatitis in patients with cystic fibrosis. Gastroenterology 140(1): 153-161,2011. PMID: 20923678.
- Ooi CYT, E. & Durie, P. Diagnostic Approach to CFTR-related Disorders, In: Cystic Fibrosis. Lung Biology in Health and Disease Series. Informa Healthcare(Allen, J.; Rubenstein, R. & Panitch, H. (Eds.)): 103-122,2010.
- Partridge AW, Melnyk RA and Deber CM. Polar residues in membrane domains of proteins: molecular basis for helix-helix association in a mutant CFTR transmembrane segment. Biochemistry 41(11): 3647-3653,2002. PMID: 11888281.
- Patrick AE and Thomas PJ. Development of CFTR Structure. Front Pharmacol 3: 162,2012. PMID: 22973227.
- Pilewski JM and Frizzell RA. Role of CFTR in airway disease. Physiol Rev 79(1 Suppl): S215-255,1999. PMID: 9922383.
- Pirot P, Eizirik DL and Cardozo AK. Interferon-gamma potentiates endoplasmic reticulum stress-induced death by reducing pancreatic beta cell defence mechanisms. Diabetologia 49(6): 1229-1236,2006. PMID: 16604358.
- Poitout V, Tanaka Y, Reach G and Robertson RP. [Oxidative stress, insulin secretion, and insulin resistance]. J Annu Diabetol Hotel Dieu: 75-86,2001. PMID: 11565471.
- Preumont V, Hermans MP, Lebecque P and Buysschaert M. Glucose homeostasis and genotype-phenotype interplay in cystic fibrosis patients with CFTR gene deltaF508 mutation. Diabetes Care 30(5): 1187-1192,2007. PMID: 17337503.
- Quinton PM. Cystic fibrosis: impaired bicarbonate secretion and mucoviscidosis. Lancet 372(9636): 415-417,2008. PMID: 18675692.
- Quinton PM. The neglected ion: HCO3. Nat Med 7(3): 292-293,2001. PMID: 11231624.
- Reid CJ, Hyde K, Ho SB and Harris A. Cystic fibrosis of the pancreas: involvement of MUC6 mucin in obstruction of pancreatic ducts. Mol Med 3(6): 403-411,1997. PMID: 9234245.
- Riordan JR. CFTR Function and Prospects for Therapy. Annual Review of Biochemistry 77(1): 701-726,2008. PMID: 18304008.
- Riordan JR, Rommens JM, Kerem BS, Alon NOA, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 245(4922): 1066-1073,1989. PMID: 2475911.
- Robertson RP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem 279(41): 42351-42354,2004. PMID: 15258147.
- Robinson J, Yates R, Harper A and Kelly C. [beta]-cells require CFTR for glucose-induced insulin secretion. Endocrine Abstracts: 34 OC32.35, 2014.
- Rosenecker J, Eichler I, Kuhn L, Harms HK and von der Hardt H. Genetic determination of diabetes mellitus in patients with cystic fibrosis. Multicenter Cystic Fibrosis Study Group. J Pediatr 127(3): 441-443,1995. PMID: 7658279.
- Rowe SM, Miller S and Sorscher EJ. Cystic fibrosis. N Engl J Med 352(19): 1992-2001,2005. PMID: 15888700.
- Rozmahel R, Wilschanski M, Matin A, Plyte S, Oliver M, Auerbach W, et al. Modulation of disease severity in cystic fibrosis transmembrane conductance regulator deficient mice by a secondary genetic factor. Nat Genet 12(3): 280-287,1996. PMID: 8589719.
- Scholte BJ, Davidson DJ, Wilke M and De Jonge HR. Animal models of cystic fibrosis. J Cyst Fibros 3 Suppl 2: 183-190,2004. PMID: 15463956.
- Schreiber R, Hopf A, Mall M, Greger R and Kunzelmann K. The first-nucleotide binding domain of the cystic-fibrosis transmembrane conductance regulator is important for inhibition of the epithelial Na+ channel. Proc Natl Acad Sci U S A 96(9): 5310-5315,1999. PMID: 10220462.
- Schwarzenberg SJ, Thomas W, Olsen TW, Grover T, Walk D, Milla C, et al. Microvascular complications in cystic fibrosis-related diabetes. Diabetes Care 30(5): 1056-1061,2007. PMID: 17322485.
- Seavilleklein G, Amer N, Evagelidis A, Chappe F, Irvine T, Hanrahan JW, et al. PKC phosphorylation modulates PKA-dependent binding of the R domain to other domains of CFTR. Am J Physiol Cell Physiol 295(5): C1366-1375,2008. PMID: 18799655.
- Seibert FS, Chang XB, Aleksandrov AA, Clarke DM, Hanrahan JW and Riordan JR. Influence of phosphorylation by protein kinase A on CFTR at the cell surface and endoplasmic reticulum. Biochimica et Biophysica Acta (BBA) - Biomembranes 1461(2): 275-283,1999. PMID: 10581361.
- Seibert FS, Linsdell P, Loo TW, Hanrahan JW, Riordan JR and Clarke DM. Cytoplasmic loop three of cystic fibrosis transmembrane conductance regulator contributes to regulation of chloride channel activity. J Biol Chem 271(44): 27493-27499,1996. PMID: 8910333.
- Sheppard DN and Welsh MJ. Structure and function of the CFTR chloride channel. Physiol Rev 79(1 Suppl): S23-45,1999. PMID: 9922375.
- Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, et al. An animal model for cystic fibrosis made by gene targeting. Science 257(5073): 1083-1088,1992. PMID: 1380723.
- Sosnay PR, Siklosi KR, Van Goor F, Kaniecki K, Yu H, Sharma N, et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet 45(10): 1160-1167,2013. PMID: 23974870.
- Stecenko AA and Moran A. Update on cystic fibrosis-related diabetes. Current Opinion in Pulmonary Medicine 16(6): 611-615,2010. PMID: 20814309.
- Stoltz DA, Meyerholz DK, Pezzulo AA, Ramachandran S, Rogan MP, Davis GJ, et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med 2(29): 29ra31,2010. PMID: 20427821.
- Stoltz DA, Meyerholz DK and Welsh MJ. Origins of cystic fibrosis lung disease. N Engl J Med 372(4): 351-362,2015. PMID: 25607428.
- Sun X, Olivier AK, Liang B, Yi Y, Sui H, Evans TI, et al. Lung phenotype of juvenile and adult cystic fibrosis transmembrane conductance regulator-knockout ferrets. Am J Respir Cell Mol Biol 50(3): 502-512,2014. PMID: 24074402.
- Sun X, Yi Y, Xie W, Liang B, Winter MC, He N, et al. CFTR Influences Beta Cell Function and Insulin Secretion Through Non-Cell Autonomous Exocrine-Derived Factors. Endocrinology 158(10): 3325-3338,2017. PMID: 28977592.
- Therien AG, Grant FE and Deber CM. Interhelical hydrogen bonds in the CFTR membrane domain. Nat Struct Biol 8(7): 597-601,2001. PMID: 11427889.
- Tucker JA, Spock A, Spicer SS, Shelburne JD and Bradford W. Inspissation of pancreatic zymogen material in cystic fibrosis. Ultrastruct Pathol 27(5): 323-335,2003. PMID: 14708723.
- van Doorninck JH, French PJ, Verbeek E, Peters RH, Morreau H, Bijman J, et al. A mouse model for the cystic fibrosis delta F508 mutation. EMBO J 14(18): 4403-4411,1995. PMID: 7556083.
- Van Goor F, Yu H, Burton B and Hoffman BJ. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J Cyst Fibros 13(1): 29-36,2014. PMID: 23891399.
- Vankeerberghen A, Wei L, Teng H, Jaspers M, Cassiman JJ, Nilius B, et al. Characterization of mutations located in exon 18 of the CFTR gene. FEBS Lett 437(1-2): 1-4,1998. PMID: 9804160.
- Veit G, Avramescu RG, Chiang AN, Houck SA, Cai Z, Peters KW, et al. From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol Bio Cell 27(3): 424-433,2016. PMID: 26823392.
- Vernon RM, Chong PA, Lin H, Yang Z, Zhou Q, Aleksandrov AA, et al. Stabilization of a nucleotide-binding domain of the cystic fibrosis transmembrane conductance regulator yields insight into disease-causing mutations. J Biol Chem 292(34): 14147-14164,2017. PMID: 28655774.
- Villalona S, Glover-Lopez G, Ortega-Garcia JA, Moya-Quiles R, Mondejar-Lopez P, Martinez-Romero MC, et al. R248G cystic fibrosis transmembrane conductance regulator mutation in three siblings presenting with recurrent acute pancreatitis and reproductive issues: a case series. J Med Case Rep 11(1): 42,2017. PMID: 28196530.
- Wang G, Linsley R and Norimatsu Y. External Zn(2+) binding to cysteine-substituted cystic fibrosis transmembrane conductance regulator constructs regulates channel gating and curcumin potentiation. FEBS J 283(13): 2458-2475,2016. PMID: 27175795.
- Wang W, El Hiani Y, Rubaiy HN and Linsdell P. Relative contribution of different transmembrane segments to the CFTR chloride channel pore. Pflugers Arch 466(3): 477-490,2014. PMID: 23955087.
- Ward CL, Omura S and Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83(1): 121-127,1995. PMID: 7553863.
- Welsh MJ and Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 73(7): 1251-1254,1993. PMID: 7686820.
- Widdicombe JH, Chen LL, Sporer H, Choi HK, Pecson IS and Bastacky SJ. Distribution of tracheal and laryngeal mucous glands in some rodents and the rabbit. J Anat 198(Pt 2): 207-221,2001. PMID: 11273045.
- Wilke M, Buijs-Offerman RM, Aarbiou J, Colledge WH, Sheppard DN, Touqui L, et al. Mouse models of cystic fibrosis: phenotypic analysis and research applications. J Cyst Fibros 10 Suppl 2: S152-171,2011. PMID: 21658634.
- Wilschanski M and Novak I. The cystic fibrosis of exocrine pancreas. Cold Spring Harb Perspect Med 3(5): a009746,2013. PMID: 23637307.
- Xu C, Bailly-Maitre B and Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest 115(10): 2656-2664,2005. PMID: 16200199.
- Yuan YR, Blecker S, Martsinkevich O, Millen L, Thomas PJ and Hunt JF. The crystal structure of the MJ0796 ATP-binding cassette. Implications for the structural consequences of ATP hydrolysis in the active site of an ABC transporter. J Biol Chem 276(34): 32313-32321,2001. PMID: 11402022.
- Zang X, Perez JJ, Jones CM, Monge ME, McCarty NA, Stecenko AA, et al. Comparison of Ambient and Atmospheric Pressure Ion Sources for Cystic Fibrosis Exhaled Breath Condensate Ion Mobility-Mass Spectrometry Metabolomics. J Am Soc Mass Spectrom 28(8): 1489-1496,2017. PMID: 28364225.
- Zhou JJ and Linsdell P. Evidence that extracellular anions interact with a site outside the CFTR chloride channel pore to modify channel properties. Can J Physiol Pharmacol 87(5): 387-395,2009. PMID: 19448737.
- Zielenski J. Genotype and phenotype in cystic fibrosis. Respiration 67(2): 117-133,2000. PMID: 10773783.
- Zoller H, Egg M, Graziadei I, Creus M, Janecke AR, Löffler-Ragg J, et al. CFTR gene mutations in pancreatitis: Frequency and clinical manifestations in an Austrian patient cohort. Wiener klinische Wochenschrift 119(17): 527-533,2007. PMID: 17943404.
- Zuelzer WW and Newton WA, Jr. The pathogenesis of fibrocystic disease of the pancreas; a study of 36 cases with special reference to the pulmonary lesions. Pediatrics 4(1): 53-69,1949. PMID: 18146464.