
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2020.16
I. The Dual Nature of the Pancreas
The pancreas is a complex gland active in digestion and metabolism through secretion of digestive enzymes from the exocrine portion and hormones from the endocrine portion. The exocrine pancreas, which accounts for more than 95-98% of the pancreas mass (43), is structurally comprised of lobules, with acinar cells surrounding a duct system. The endocrine pancreas makes up only 2% of the pancreatic mass and is organized into the islets of Langerhans— small semi-spherical clusters of about 1500 cells (73) dispersed throughout the pancreatic parenchyme— which produce and secrete hormones critical for glucose homeostasis. The existence of islets was described by Paul Langerhans in 1869, and the functional role of islets in glucose homeostasis was first demonstrated in 1890 when Joseph von Mering and colleagues showed that dogs developed diabetes mellitus following pancreatectomy (22). Though islet mass may vary between individuals—an example is the increase in the setting of adult obesity (83)— the average adult human pancreas is estimated to contain one to two million islets (33, 94). In humans, the concentration of islets is up to two times higher in the tail compared to the head and neck. However, the cellular composition and architectural organization of cell types within the islets is preserved throughout the pancreas (103).
Each pancreatic islet is composed of α, β, d, ε and PP (F) cells; these are primarily endocrine (hormone-secreting) cells, containing numerous secretory granules with stored hormone molecules, ready for release upon receipt of the appropriate stimulus. Insulin-producing b cells are the most common cell type, making up 50-70% of islet mass, with small islets containing a greater percentage of β-cells in contrast to moderate or large islets (4,5). b cells were first discovered in 1907 by silver staining (50) and were the second islet cell type discovered, thus designated “b”-cells. In addition to insulin, b cells also produce islet amyloid polypeptide (IAPP), or amylin, which is packaged and released within insulin-containing granules (44). Amylin reduces post-prandial hyperglycemia by slowing gastric emptying and promoting satiety.
Glucagon-producing a cells were discovered before b cells, by alcohol fixation, thereby garnering their name “a” –cells (50). As the second most abundant islet cell type, they make up about 35% of islet mass in humans (8) but less in rodents. Glucagon’s primary function is to prevent hypoglycemia by stimulating glycogenolysis and hepatic gluconeogenesis (6). Somatostatin-producing d cells comprise less than 10% of islet mass, and are evenly distributed throughout the pancreas (1). Somatostatin is an inhibitory peptide hormone, inhibiting both endocrine and gastrointestinal hormones. Pancreatic polypeptide (PP) producing cells, also known as Ɣ or "F" cells (43, 79), comprise less than 5% of islet mass, and like a cells, are most prominent in the head of the pancreas. PP has roles in exocrine and endocrine secretion functions of the pancreas (107). Ghrelin-producing e cells are the last discovered islet endocrine cell type. Although present in islets, ghrelin is predominately produced in the stomach; ghrelin suppresses insulin release, and plays a role in regulating energy homeostasis (101).
The close proximity of the acini and the islets of Langerhans mirrors their functional interplay. The anatomic structure of the pancreatic parenchyme allows for a paracrine effect of the islet hormones on adjacent acinar cells, termed the ‘islet-acinar’ axis (2, 108). Notably, the islets are highly vascularized—receiving 15% of pancreatic arterial blood flow despite composing only 2% of the pancreatic mass (41). Via the islet-acinar portal system, blood bathing the pancreatic islets flows into a capillary bed within the pancreatic acini, thus exposing the acinar pancreas to the islet hormones (66). Insulin binds to an insulin receptor on acinar tissue and potentiates amylase secretion (109). In contrast, somatostatin inhibits pancreatic exocrine secretion (64); endogenous PP is also largely noted to inhibit pancreatic exocrine secretion (90, 107). Studies have been inconsistent with regards to the effect of glucagon, some suggesting a stimulatory effect while many suggesting an inhibitor effect of glucagon on secretion of zymogen granules (2).
II. Insulin Structure
The hormone insulin was first isolated in the 1920’s by Dr. Frederick Banting and a medical student Charles Best, garnering Banting (jointly with John James Rickard Macleod) the Nobel Prize in Medicine in 1923. This was a critical step forward in diabetes care, as porcine insulin therapy was then made available for human use to treat type 1 diabetes, an otherwise fatal disease. In the 1950’s Frederick Sanger determined its primary amino acid structure, consisting of an A and a B chain connected by disulfide bonds (40, 84). Ten years following this discovery, these chains were found to be from the same polypeptide precursor, preproinsulin. In the 1960’s Dorothy Hodgkin defined its tertiary structure. During translation of preproinsulin from its mRNA, the N-terminal signal peptide is cleaved to yield proinsulin. The proinsulin molecule is a single chain polypeptide containing both the A-chain (21 amino acids long) and the B-chain (30 amino acids long). In proinsulin, two chains are connected by C-peptide, which is cleaved to release C-peptide and the remaining insulin molecule, which contains the A- and B-chains connected via two disulfide bonds (40). Although insulin and C-peptide are co-released from b cell secretory vesicles into circulation (81), only insulin is biologically active in regulating blood glucose. C-peptide, however, can serve as a useful clinical and research measure of endogenous insulin production, in patients receiving exogenous insulin injections.
III. Insulin Gene Transcription
The insulin gene on chromosome 11 is primarily expressed in pancreatic b cells, but is expressed in low levels in the brain, thymus, and in the yolk sak during fetal development (28, 52, 72). It has three exons and two introns, and its transcription results in the 446 base pair preproinsulin mRNA (Figure 1).
Figure 1. Various levels of glucose regulation of insulin gene expression. Glucose stimulates nuclear translocation of Pdx-1; promotes Pdx-1 and MafA phosphorylation and binding to the insulin promoter; and stimulates transcription of the insulin gene, pre-mRNA splicing, translation, and mRNA stability. (Used with permission from (74)).
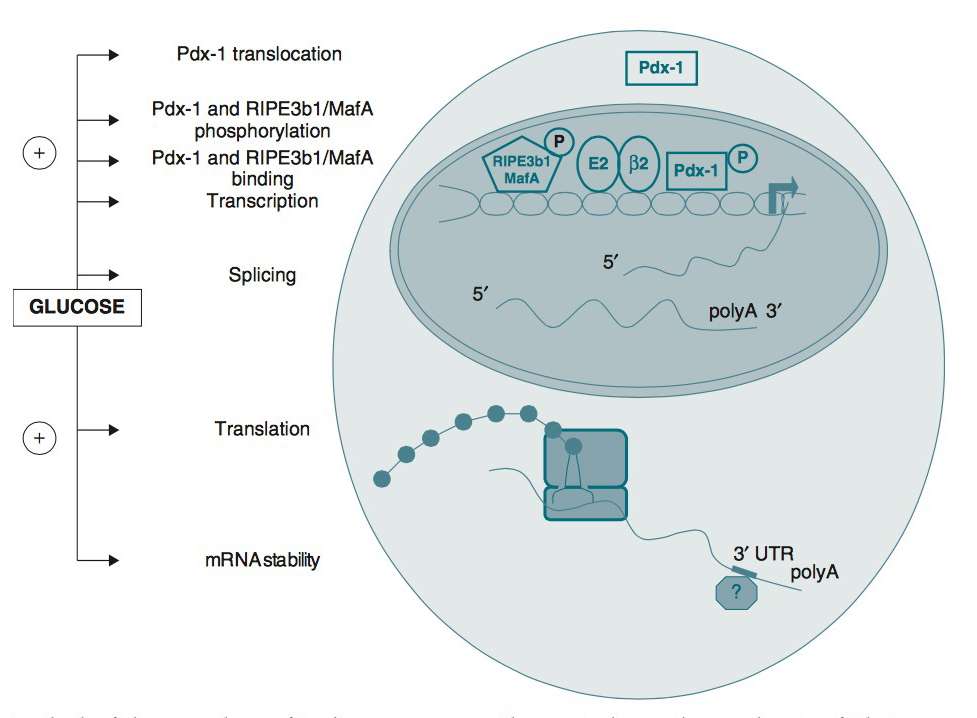
Transcription of the insulin gene to preproinsulin mRNA is sophisticated and reflects the tight regulation by transcription factors and recruited coactivators. Pdx-1, NeuroD1 and MafA are important transcription factors in b cell function, respond to elevated glucose levels. Individual b cells respond to ambient glucose with differential insulin secretion, and these changes are apparent at the level of gene transcription (16). At the level of the islet, rapid increase in blood glucose results in rapid elevation in preproinsulin mRNA in the endocrine pancreas. A rapid decrease in blood glucose results in a slow decline in preproinsulin mRNA.
This is due to the unusual stability of preproinsulin mRNA, further stabilized by increased glucose concentrations (25). The specific regulation of this molecule’s translation is the primary mechanism of insulin production control (74).
Mature insulin-containing granules are retained from a few hours up to several days within the b cell, ready for transport to plasma membrane and exocytosis when stimulated. The storage of insulin in mature b granules is far greater than that secreted (58, 80). During a 1 hour glucose stimulation only ~1-2% of insulin within a primary islet b cell is released (102). The insulin content within a given b cell remains relatively constant in the short term, but in the long term will adapt in response to physiologic demands (102).
IV. Insulin Function
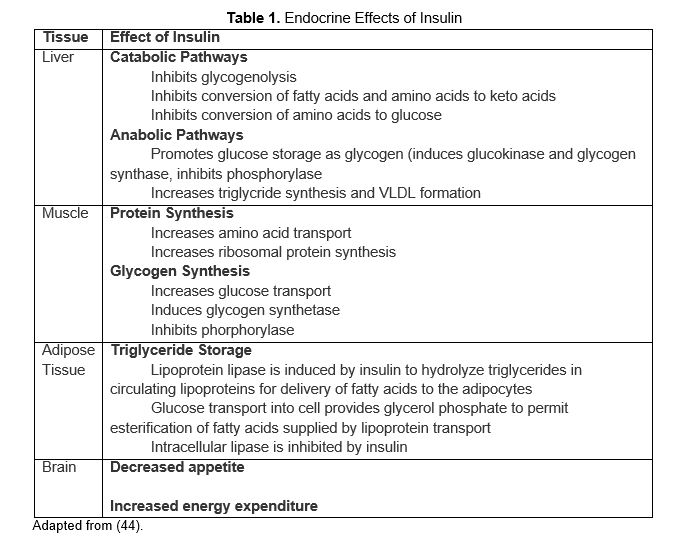
In an evolutionary milieu of sporadic access to nutrients, insulin became critical in facilitating survival. As an anabolic hormone, insulin controls metabolism of carbohydrates, lipids, and protein. It mediates the availability of energy sources in both fasting and fed states. Insulin promotes energy storage in the fasting state and energy utilization and uptake in the fed state (Table 1). In so doing, it maintains serum glucose levels within a narrow physiologic range despite variation in energy intake and expenditure. Insulin acts at extracellular insulin receptors in multiple organ tissues including the liver, muscle, and adipose tissue (43), and its effect depends on interstitial insulin concentration which is influenced by insulin secretion rate from b cells and clearance from circulation (68).
The liver serves as the primary storage site for glucose, accounting for 80% of glucose production in fasting states with the kidney only contributing 20% (18, 96).
Table 1. Endocrine Effects of Insulin. (Adapted from Masharani and German (60)).
To preserve glucose stores, the low insulin concentrations in the portal venous blood—as seen in the fasting state-- allows minimal glucose production, only enough to match the needs of essential glucose-dependent tissues including the red blood cells and the central and peripheral nervous systems. The liver also clears insulin more rapidly in the fasting state, thus maintaining low circulating insulin levels. Low insulin concentrations also contribute to lipolysis in adipocytes, releasing free fatty acids to encourage utilization of lipid over glucose to meet resting energy needs. Hepatic glucose release during fasting states through glycogenolysis and gluconeogenesis is stimulated by counter-regulatory, or ‘anti-insulin’ hormones. Glucagon plays a major role, with synergistic effects from catecholamines, cortisol, and growth hormone (68).
By contrast, in the fed state-- in response to digestion and absorption of nutrients-- circulating insulin concentration increases in the portal vein secondary to insulin secretion from pancreatic b cells. The increased insulin and glucose concentrations normally limit hepatic glucose production and stimulate liver glucose uptake through glycogen deposition (23, 32, 91). Insulin causes upregulation of hexokinase, phosphofructokinase, and glycogen synthase within hepatocytes, thus inhibiting glycogenolysis and gluconeogenesis and stimulating glycogen synthesis (18).
The effect of insulin on gluconeogenesis can be direct (via its effect on the liver) or indirect via its effect on islet a cells (by decreasing glucagon secretion), adipose tissue (by suppressing lipolysis), skeletal muscle (by reducing proteolysis), and the brain (pleiotropic effect) (32,
65).
In situations when there is poor insulin response such as type 2 diabetes mellitus or insulin resistance, the process of gluconeogenesis continues even in the fed state, thus, further compounding hyperglycemia (32).
Liver clearance of insulin is decreased in the fed state, thus further increasing the circulating insulin concentration. In adipocytes, insulin upregulates lipoprotein lipase and downregulates hormone sensitive lipase, which inhibits lipolysis and subsequent free fatty acid release (29). In hepatocytes, insulin instead stimulates hepatic free fatty acid synthesis from glucose, thereby increasing lipid stores. Proteolysis of skeletal muscle is also inhibited by insulin, which along with lipolysis inhibition, limits delivery of glucose precursors (glycerol and amino acids) to the liver. Systemic circulation of insulin stimulates glucose uptake and utilization in skeletal muscle and adipocytes.
In summary, the release of insulin in the fed state, (1) promotes accumulation of energy stores through glycogenesis and lipogenesis, (2) reduces new hepatic glucose output by preventing glycogenolysis and gluconeogenesis (in the non-insulin resistant, non-diabetic individual), and (3) promotes uptake of glucose by skeletal muscle and fat, the net effect of which is to maintain a normal circulating serum glucose levels while storing extra energy for use during later periods of fasting (Figure 2).
Figure 2. Glucose homeostasis in the fed state. Glucose absorbed from the digestive tract enters the portal blood flow and then systemic circulation. In the fed state, increased glucose stimulates insulin release from the pancreatic β-cells. Insulin acts at the level of the liver to inhibit hepatic gluconeogenesis, at the skeletal muscle to promote storage of glucose as glycogen, and in the adipocytes to stimulate lipogenesis. High insulin levels inhibit the release of non-esterified fatty acids. Incretin hormones released from small intestine in response to a meal augment pancreatic glucose-stimulated insulin secretion. Brain and red blood cells take up glucose independently of insulin in the fasting and fed state. In the fasting state (not shown), in the setting of low circulating insulin, hepatic gluconeogenesis, glycogenolysis, and release of non-esterified fatty acids occurs. Solid line stimulation; dashed lines denote inhibition.

Glucose movement into cells is made possible by specific protein transporters within the plasma membrane of glucose-responsive cells that reversibly bind glucose and transport it bidirectionally across the cell membrane. There are 14 known glucose transporters (GLUTs) (56, 99). They are present in different concentrations and in different tissues, with varying sensitivity to insulin (Table 2).
Table 2. Most common glucose transporters (GLUT) in human tissues.
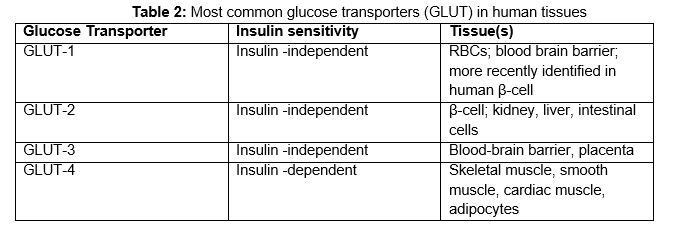
Tissues such as muscle and adipocytes carry the insulin-dependent glucose transporter GLUT-4 and uptake of glucose into these tissues occurs only under conditions of adequate circulating insulin. In contrast, vital organs such as red blood cells, brain, placenta, and kidney carry insulin-independent glucose transporters. Thus, these latter essential organs can continue to function even in states of insulin deficiency. β-cells also depend upon on a glucose-independent transporter, GLUT2, to allow ambient blood glucose to freely transverse the β-cell membrane in order to stimulate insulin production.
V. Insulin Secretory Pathway
The pancreatic b-cells act as a self-contained system to secrete insulin in response to changes in ambient blood glucose concentration, in order to maintain glucose homeostasis. Glucose is freely taken up into the b-cell via GLUT transporters, metabolized to produce ATP, which triggers a cascade of signals within the b cell necessary for glucose-induced insulin secretion. While GLUT2 has been traditionally assumed as the major mediator of glucose uptake into b-cells based on extrapolation from rodent studies and subsequent confirmation of GLUT2 transporters on human β-cells (17, 71, 100), more recent studies in human islets suggest that the other insulin-independent glucose transporters GLUT1 and GLUT3 play a more important role, and are the main glucose transporters in human islet β-cells (3, 98). This redundancy explains why individuals with variants in the gene encoding GLUT2 (SLC2A2 mutations, or Fanconi–Bickel syndrome) do not have significant abnormalities in insulin secretion (89).
As blood glucose increases (e.g., after a meal), there is a resultant flux of glucose across the GLUT transporters in the β-cell. Subsequently, within the β-cell, glucose is phosphorylated to glucose-6-phosphate by glucokinase. This is the rate-limiting step of insulin secretion, and as such, glucokinase is considered the “glucose sensor” for the b-cell (17, 61). Because of this critical role of glucokinase, individuals with heterozygous mutations in the glucokinase gene have a mild to moderate non-progressive hyperglycemia (maturity onset of diabetes in the young, type 2) (12). Once in the mitochondria, glucose-6-phosphate is metabolized by the Krebs cycle to produce ATP. The resultant ATP binds and closes the ATP-dependent potassium channel, a pore across the cell membrane, which consists of four Kir6.2 subunits and has four regulatory SUR (sulfonylurea receptor) subunits. Channel closure blocks potassium exit from the b-cell, thus depolarizing the cell membrane.
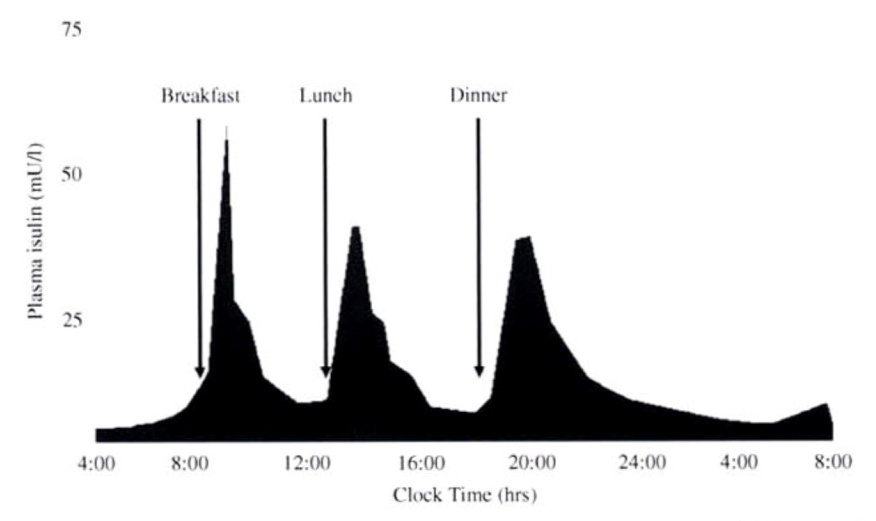
Once the cell is depolarized, the L-type voltage-gated calcium channels are triggered, increasing influx of calcium and resultant cellular calcium concentrations. Increased cytoplasmic calcium concentrations triggers release of insulin and C-peptide from a pool of insulin-containing docked secretory vesicles and stimulates the migration of additional vesicles to the cell membrane (Figure 3). Though simple glucose-stimulated insulin secretion (GSIS) as described above is considered the primary pathway for insulin secretion, the full picture is more nuanced. GSIS is augmented by amplifying pathways including: (1) metabolic amplification by amino acids, free fatty acids, and glucose itself; and (2) neurohormonal amplifiers such as GLP-1 and parasympathetic innervation (14, 34, 48, 76). More recent data from mice suggest a role for skeletal muscle in regulating b-cell insulin secretion via production of an anorexic factor typically derived from the hypothalamus in the brain called BDNF (brain-derived neurotrophic factor) (26). This effect is mediated via the BDNF receptor (TrkB.T1) which is expressed on b-cells, and is thought to play a potential role in exercise-induced glucose metabolism (26). These physiologic, and pharmacologic, triggers for insulin secretion are further described in the following sections. About half of insulin secretion occurs as basal insulin release, while the other half occurs as ‘bolus’ insulin responses to a meal (62). This basal-bolus dynamic of insulin secretion is important in considering clinical management of the patient with diabetes (Figure 4). In those with complete insulin deficiency—e.g. type 1 diabetes, late-stage type 2 diabetes, or late-stage chronic pancreatitis diabetes—insulin analogs are administered by multiple daily injections or a continuous subcutaneous insulin infusion (insulin pump) to mimic this basal-bolus pattern of endogenous insulin secretion.
Figure 3. Glucose stimulated insulin-secretion coupling in the b cell. The main pathway of glucose stimulated insulin secretion in the beta cell. Glucose enters the beta cell through GLUT transporters. Glucose metabolism results in an enhanced cytoplasmic ATP/ADP ratio which prompts closure of ATP-sensitive K+ (KATP) channels in the plasma membrane evoking membrane depolarization and subsequent opening of voltage-gated Ca2+ channels. This culminates in an increase in cellular Ca2+ influx- a primary driver of insulin exocytosis. Ca2+ and vesicle docking and fusion events can also be modulated by agents acting through the phospholipase C (PLC)/protein kinase C (PKC) or adenylate cyclase (AC)/protein kinase A (PKA) pathways, via neuro-hormonal and metabolic amplification (not illustrated). (Used with permission from (59)).

Figure 4. Diagrammatic illustration of insulin secretion. A low background secretion exists upon which is superimposed insulin secretory bursts stimulated by food intake. (Used with permission from Thompson, Christie, and Hindmarsh (97)).
VI. Regulation of Insulin Release
Insulin release from pancreatic b cells is tightly regulated, and allows the sensitive response of insulin levels to calorigenic nutrients in the body. Glucose, free fatty acids, and amino acids serve as fuel stimuli for insulin release, promoting insulin granule exocytosis. Additional hormonal factors influence the regulation pathway. Pharmacological agents can also be used to augment insulin release.
A. Glucose-stimulated insulin secretion
Glucose-stimulated b-cell insulin release is the primary mechanism of insulin regulation (Figure 3) (35, 88).In humans, this is illustrated by use of the hyperglycemic clamp technique (Figure 5), in which individuals are made rapidly hyperglycemic by injection of intravenous dextrose, and hyperglycemia is maintained by variable rate dextrose infusion at a predefined target glucose (20). Hyperglycemic clamp studies demonstrate a dose-response of insulin secretion in response to glucose concentration, with greater degrees of hyperglycemia eliciting a more robust insulin secretory response in the non-diabetic individual (70, 82). Using this research technique, two distinct phases of insulin secretion are observed. During the first phase insulin response (otherwise referred to as the acute insulin response to glucose, AIRglu), there is an immediate and transient rise in insulin secretion, peaking by five minutes and lasting no more than ten minutes. This first phase of insulin secretion is hypothesized to largely result from the immediate release of insulin from insulin secretory vesicles that are already docked and primed at the β-cell membrane. This first phase response is lost under conditions of diabetes mellitus, when β-cell reserves are exhausted (104). The second sustained phase begins at this ten-minute time-point and lasts as long as the glucose elevation is elevated. The second phase results from recruitment of insulin secretory vesicles to the β-cell membrane, and is also controlled by intracellular calcium levels (68).
Figure 5. Hyperglycemic clamp illustration. Example of hyperglycemic clamp testing in obese adolescents with normal glucose tolerance (NGT, solid line), impaired glucose tolerance (IGT, dashed line), and type 2 diabetes (T2DM, dotted line). In the hyperglycemic clamp in healthy, non-diabetic individuals, glucose concentration is briskly elevated by administering a suitable intravenous glucose infusion at time 0. This elicits a rapid and short-lived insulin secretion peak (first-phase secretion) due to release of preformed insulin vesicles, followed by a drop towards basal levels and then by a relatively rapid return to a sustained increase in insulin in the second half of the clamp (second-phase secretion) as dextrose infusion is continued. This example illustrates the loss, in first and second phase insulin secretion, as individual progress from normal to impaired glucose tolerance, to type 2 diabetes. In the latter, the first phase insulin response is essentially lost and the second phase insulin response is reduced. (With permission from Wiess et al, (104)).
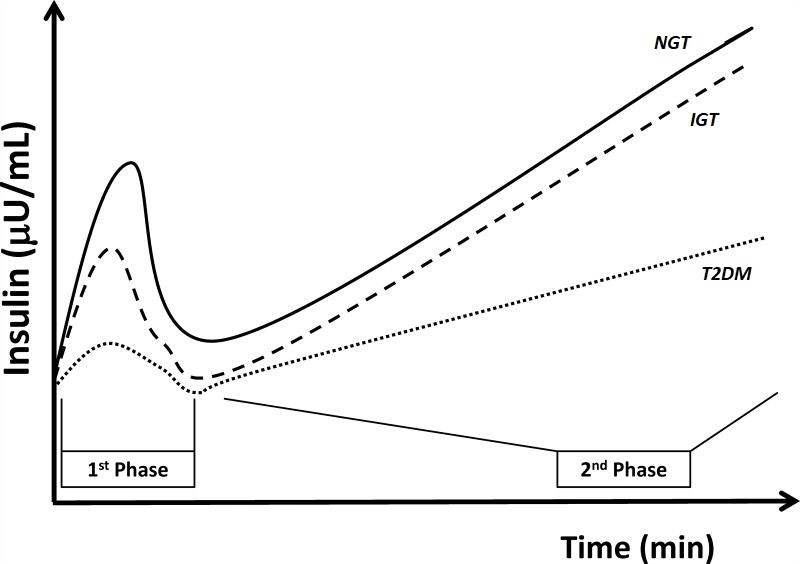
It is unclear how significant first and second-phase insulin responses are in the ‘real world’ setting. In contrast to this scenario of rapid infusion of intravenous glucose, ingestion of a physiologic meal results in a much more gradual rise of serum glucose (15). However, characterization of first phase insulin response is critically important in diabetes research. In progression to type 1 and type 2 diabetes mellitus, the earliest abnormality is a loss in the first phase insulin secretion (measured as the AIRglu). Although chronic pancreatitis diabetes is much less studied, this appears likely also to be the case in patients with chronic pancreatitis progressing to diabetes based on limited studies, and often in patients with chronic pancreatitis who have diabetes or pre-diabetes (18). The AIRglu can be elicited experimentally by administering a 0.3 g/kg dextrose bolus and sampling insulin levels at baseline and at +2, 3, 4, 5, 7 and 10 min after the rapid IV administration of dextrose. The AIRglu can be calculated using various methods, including but not limited to the area under the curve minus baseline or mean of the 2-5 min values minus baseline.
Interestingly, glucose also appears to be a ‘metabolic amplifier’ for insulin secretion, in addition to the classic pathway of glucose-stimulated insulin secretion. Glucose amplifies insulin secretion, a process called time dependent potentiation of insulin secretion—whenβ-cells are exposed to hyperglycemia this augments subsequent insulin secretory responses to glucose (112).
B. Proteins and Amino Acids
Pancreatic b cells adjust insulin secretion based on other nutrients including amino acids, fatty acids, and ketone bodies. Oral protein intake, and subsequent rise in serum amino acids, stimulate insulin release by direct b cell stimulation (11, 45, 69). The insulinotropic effect varies among amino acids, and there appears to be a synergistic effect of mixed amino acids versus individual administration (24).
Some amino acids stimulate insulin secretion by acting as substrates in the Krebs cycle, metabolizing glucose-6-phosphate to create ATP. Enzymes active in b cell mitochondrial amino acid metabolism have been implicated in hyperinsulinemic hypoglycemic syndromes associated with high-protein containing meals (Prentki, Matschinsky, and Madiraju 2013). The ATP binds to and closes the potassium channel, leading to cell depolarization and insulin secretion. There seems to be a direct effect of proteins and amino acids on b cell glucose sensitivity, because ingestion of amino acids with glucose results in the same plasma insulin concentrations as elicited by a lower level of glucose alone (27).
C. Lipids and Free Fatty Acids
It is generally accepted that lipids play a role in insulin secretion signaling, but the precise pathways and molecules involved in the process remain less well understood. Lipid breakdown and metabolism to signaling molecules has been linked to glucose metabolism through enhanced membrane phospholipid metabolism turnover and other pathways yet to be firmly established. It is thought that free fatty acids (FFA) modulate β-cell insulin secretion either directly via GPR40 (G-protein coupled receptor on the β-cell) leading to insulin secretion, or indirectly via oxidation of FFA to acyl coA, which enters the Krebs cycle and generates ATP (43).
Glucose and FFA metabolism have been shown to be tightly linked and likely includes malonyl-CoA/carnitine palmitoyltransferase I/fatty acyl-CoA metabolic signaling network and the glycerolipid/free fatty acid (GL/FFA) cycle (13, 75). The GL/FFA cycle along with the Krebs cycle and pyruvate cycling are the three likely interlined metabolic cycles that play essential roles in insulin secretion promoted by glucose, FFA, and amino acids (76). In so doing, FFA work synergistically with glucose-stimulated insulin secretion to enhance insulin secretion in nutrient-abundant states.
Chronic elevation of fatty acids may increase basal insulin secretion levels but inhibits glucose-stimulated insulin secretion. Chain length and degree of saturation affect the role free fatty acids play in regard to insulin plasma levels (95). Adipose tissue responds to insulin resistance with a persistently elevated rate of lipolysis, thus increasing the plasma free fatty acid levels. This is believed to be important in type 2 diabetes development (46).
D. Incretin Hormones
Though glucose concentrations can account for the majority of changes in insulin concentrations, complex studies evaluating in vivo insulin concentrations following meals have identified other factors (67). Indeed, insulin secretion following an oral glucose tolerance test is directly related to blood glucose levels, but is considerably higher than predicted following intravenous glucose infusion. These findings suggest a role for potentiating effects on insulin release by hormones that specifically respond to oral glucose, the “incretin effect”. This terminology is derived from intestinal hormones called incretins, which are credited with facilitating this response.
The most active incretins are glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) (38, 106), but gastrin, secretin, and cholecystokinin may also have minor roles. In response to glucose and other nutrients, intestinal L cells secrete GLP-1 and K cells secrete GIP. These hormones then bind their specific receptors on the pancreatic b-cell membrane. GLP-1 binds a G protein-coupled receptor. This results in direct activation of insulin gene’s cyclic-AMP (cAMP) response element (CRE) of the 5’-proximal control sequence (49, 93). It can also augment Pdx-1 binding in the setting of a glucose-stimulus, and stimulate transcription of the PDX-1 gene (39). Finally, it potentiates glucose-induced insulin gene transcription by activating NFAT (nuclear factor of activated T-cell) (57). The incretin effect is also mediated by glucose concentration, stimulating more insulin secretion in more extreme hyperglycemic states. GIP and GLP-1 receptors also exist on neuronal cells (e.g. nodose ganglion of the Vagus nerve), suggesting an additional indirect role in b-cell regulation (37). GLP-1 and GIP are cleared by dipeptidyl peptidase-4 (DPP-4) which is present on vascular endothelium. As a result, their half-life iin the circulation is 2-3 minutes and 4-5 minutes, respectively (63).
VII. Insulin’s Counterregulatory Hormones
The tight control of energy utilization and stores by insulin is balanced by the counterregulatory hormones glucagon, pancreatic polypeptide, somatostatin, cortisol, catecholamines, and growth hormone. There is asymmetry in the glucose regulation hormones, as insulin is the only hormone to prevent against hyperglycemia, while at least three other hormones (cortisol, glucagon, and adrenaline) prevent hypoglycemia. Collectively, these counter-regulatory hormones act to promote glucose release from the liver by glycogenolysis and gluconeogenesis, and inhibit glucose storage during times of starvation.
Glucagon is formed within pancreatic islet cells and has a hyperglycemic effect on the body (6). Its name is derived from glucose agonist (36). Glucagon carries out its effects via activating its G-protein coupled receptor that is found in various organs/tissues such as the liver, adipose tissue, kidneys, gastrointestinal (GI) tract, brain, and islet a- and b-cells (105). It stimulates glucose production from amino acids and glycerol through gluconeogenesis and from the liver through glycogenolysis. Glucagon also acts at the adipocyte to upregulate hormone-sensitive lipase, thereby enhancing lipolysis and free fatty acid delivery to the liver (54). In the brain it increases satiety (9), and in the GI tract it reduces GI motility (47). Glucagon, via its autocrine role, stimulates further glucagon secretion through its effect on a-cells (55). Interestingly, glucagon stimulates insulin secretion via glucagon’s effect on b-cells. It is not clear if this effect is mediated mainly via glucagon’s effect on glucagon receptors or on GLP-1 receptors (105). This effect on insulin secretion occurs in the fed state (10).
Mechanisms explaining glucagon secretion are not as well understood as those of insulin secretion, although the direct effect of reduced glucose on cAMP (111), and the sodium-glucose cotransporters (SGLT) are thought to play a role in a-cell glucose transport (3). Mice and human data suggest that a-cell inhibition can occur, at least in part, due to the paracrine action of somatostatin from d-cells as a result of gap junction-dependent activation by adjacent b-cells (7).
Cortisol antagonizes insulin’s function by promoting protein catabolism to provide amino acid substrate for gluconeogenesis and also impairs peripheral insulin-mediated glucose uptake.
Catecholamines directly affect b-cell secretion of insulin, as activation of a-2 inhibits insulin secretion and b stimulation increases it. Catecholamines promote adipocyte lipolysis, hepatic glycogenolysis and peripheral insulin resistance. Epinephrine inhibits insulin secretion through inhibiting the rate of insulin gene transcription (110). Somatostatin also destabilizes the preproinsulin mRNA, resulting in premature degradation (72).
Somatostatin is released from pancreatic islet d cells and exerts inhibitory effect on pancreatic b cells. Once bound to specific somatostatin receptors, b cell membrane repolarization is induced, resulting in reduction of calcium influx and thereby inhibiting insulin release (88, 110).
Pancreatic polypeptide (PP) is secreted by PP, or F, cells in pancreatic islets (107). In addition to its effects reducing gastric acid secretion, decreasing gastric emptying and slowing upper intestinal motility, PP acts within the pancreas to self-regulate pancreatic insulin secretion.
VIII. Pharmacologic Modulators of Insulin Response
There is a plethora of pharmcologic agents designed to target various aspects of glucose metabolism. In this chapter, we provide examples of pharmacologic agents that directly or indirectly modulate insulin response.
A. Incretin mimetics
Diabetes therapeutics have recently utilized the role of incretin hormones for pharmacologic benefit. Due to the desirable effect of GLP-1 on hemoglobin A1c (HbA1c) reduction and weight loss (42), GLP-1 receptor agonists and inhibitors of its degradation via dipeptidyl peptidase-4 (DPP-4) inhibitors, have been used to treat type 2 diabetes since 2005.
Short-acting GLP-1 receptor agonists (such as exenatide and Liraglutide), and long-acting GLP-1 receptor agonists (such as weekly exenatide and Semaglutide) potentiate insulin secretion and reduce gastric motility (31). Given that GLP-1 receptor agonists potentiate glucose-induced insulin gene transcription, they, alone, do not induce hypoglycemia when used as monotherapy (21,79).
DPP-4 inhibitors (such as sitagliptin) can significantly increase the peak post-prandial concentration of GLP-1 (Herman et al. 2006). Additionally, sitagliptin has been found to potentiate GSIS independently of GLP-1 via islet peptide tyrosine tyrosine (PYY) (30).
B. Sulfonylureas
Through a direct action on pancreatic islet cells, sulfonylureas are pharmacological agents that stimulate insulin secretion, thereby lowering blood glucose levels. This class of medication was discovered by happenstance in 1942 when Marcel Janbon, a clinician at the Clinic of the Montpellier Medical School in France found his patients treated for typhoid fever with a new sulfonamide (2254 RP) developed hypoglycemia. Shortly after this, his colleague Professor August Loubatieres established the hypoglycemic property of 2254 RP and its analogues were by direct action on pancreatic islets. This marked the birth of sulfonylureas for treatment of certain forms of diabetes (57). It was not until 50 years later that the mechanism of action was discovered. Sulfonylurea was found to bind to and block the potassium ATP channel on the b-cell surface, thus depolarizing the membrane and provoking calcium influx, raising intracellular calcium concentration, and triggering insulin secretion (86, 87). Sulfonylurea binding to the sulfonylurea receptor associated with the K-ATP channel stimulates events similar to those in response to glucose stimulation.
Sulfonylureas are also used in the chronic treatment of type 2 diabetes mellitus for both their effects on insulin release and blood glucose reduction. In contrast to acute use of sulfonylureas, chronic use results in improved blood glucose control, but with less rather than more insulin secretion (78). Assessments of its chronic effects are difficult to interpret, given that the magnitude of sulfonylurea stimulation of insulin secretion are multifactorial (53).
C. Insulin Sensitizers
Biguanides (such as metformin) and Thiazolidenediones (such as pioglitazone) improve hepatic and peripheral (muscle and fat tissue) insulin sensitivity, respectively. Metformin is by far the most widely used pharmacologic agent as first line therapy in patients with type 2 diabetes mellitus. Similar to thiazolidenediones, metformin has an effect on improving peripheral insulin sensitivity in addition to reducing hepatic glucose output. Contrary to thiazolidenediones and sufonylureas, metformin does not cause weight gain, and in fact, it has a modest weight loss effect. When used as monotherapy, metformin does not induce hypoglycemia (85).
D. Diazoxide
Diazoxide is a sulfonamide pharmacological agent used in treatment of hyperinsulinism, insulinoma, and hypoglycemia due to overtreatment with sulfonylureas. It works by opening b cell membrane potassium ATP channels, hyperpolarizing the b cells, thus decreasing intracellular calcium concentration and inhibiting insulin secretion (27).
IX. Conclusion
In conclusion, although the pancreatic islets comprise only a small portion of the pancreas, pancreatic islets play a vital role in our well-being and survival through control of glucose homeostasis. Most critically, loss of insulin production from the pancreatic β-cells, whether due to autoimmune destruction in type 1 diabetes mellitus, exhaustion and genetic predisposition to failure in type 2 diabetes mellitus, or bystander fibrotic destruction in pancreatic exocrine disease, results in diabetes. Insulin secretion is tightly regulated in healthy non-diabetic individuals, with both insulin gene transcription and exocytosis from insulin-containing granules responsive to rises in ambient circulating blood glucose. Other nutrients (protein and lipid) play a smaller role. In contrast, the other pancreatic islet cells, particularly the glucagon-producing α cells, play a key role in glucose counter-regulation to avoid dangerous hypoglycemia.
X. References
- Baetens D, Malaisse-Lagae F, Perrelet A and Orci L. Endocrine pancreas: three-dimensional reconstruction shows two types of islets of langerhans. Science 206(4424): 1323-1325, 1979. PMID: 390711.
- Barreto SG, Carati CJ, Toouli J and Saccone GT. The islet-acinar axis of the pancreas: more than just insulin. Am J Physiol Gastrointest Liver Physiol 299(1): G10-22, 2010. PMID: 20395539.
- Berger C and Zdzieblo D. Glucose transporters in pancreatic islets. Pflugers Arch 472(9): 1249-1272. 2020. PMID: 32394191.
- Bonner-Weir S, Sullivan BA and Weir GC. Human Islet Morphology Revisited: Human and Rodent Islets Are Not So Different After All. J Histochem Cytochem 63(8): 604-612, 2015. PMID: 25604813.
- Bosco D, Armanet M, Morel P, Niclauss N, Sgroi A, Muller YD, et al. Unique arrangement of alpha- and beta-cells in human islets of Langerhans. Diabetes 59(5): 1202-1210, 2010. PMID: 20185817.
- Bozadjieva N, Williams JA, and Bernal-Mizrachi E. Glucagon. Pancreapedia: Exocrine Pancreas Knowledge Base, 2013. DOI: 10.3998/panc.2013.23.
- Briant LJB, Reinbothe TM, Spiliotis I, Miranda C, Rodriguez B, and Rorsman P. 2018. δ-cells and β-cells are electrically coupled and regulate α-cell activity via somatostatin. J Physiol 596(2): 197-215, 2018. PMID: 28975620.
- Brissova M, Fowler MJ, Nicholson WE, Chu A, Hirshberg B, Harlan DM, et al. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J Histochem Cytochem 53(9): 1087-1097, 2005. PMID: 15923354.
- Campbell JE and Drucker DJ. Islet α cells and glucagon--critical regulators of energy homeostasis. Nat Rev Endocrinol 11(6): 329-338, 2015. PMID: 25850661.
- Capozzi ME, Wait JB, Koech J, Gordon AN, Coch RW, Svendsen B, et al. Glucagon lowers glycemia when β-cells are active. JCI Insight 5(16): e129954, 2019. PMID: 31335319.
- Carr RD, Larsen MO, Winzell MS, Jelic K, Lindgren O, Deacon CF, et al. Incretin and islet hormonal responses to fat and protein ingestion in healthy men. Am J Physiol Endocrinol Metab 295(4): E779-784, 2008. PMID: 18612044.
- Chakera AJ, Steele AM, Gloyn AL, Shepherd MH, Shields B, Ellard S, et al. Recognition and Management of Individuals With Hyperglycemia Because of a Heterozygous Glucokinase Mutation. Diabetes Care 38(7): 1383-1392, 2015. PMID: 26106223.
- Chen S, Ogawa A, Ohneda M, Unger RH, Foster DW and McGarry JD. More direct evidence for a malonyl-CoA-carnitine palmitoyltransferase I interaction as a key event in pancreatic beta-cell signaling. Diabetes 43(7): 878-883, 1994. PMID: 8013751.
- Curi R, Lagranha CJ, Doi SQ, Sellitti DF, Procopio J, Pithon-Curi TC, et al. Molecular mechanisms of glutamine action. J Cell Physiol 204(2): 392-401, 2005. PMID: 15795900.
- Curry DL, Bennett LL and Grodsky GM. Dynamics of insulin secretion by the perfused rat pancreas. Endocrinology 83(3): 572-584, 1968. PMID: 4877098.
- de Vargas LM, Sobolewski J, Siegel R and Moss LG. Individual beta cells within the intact islet differentially respond to glucose. J Biol Chem 272(42): 26573-26577, 1997. PMID: 9334237.
- De Vos A, Heimberg H, Quartier E, Huypens P, Bouwens L, Pipeleers D, et al. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J Clin Invest 96(5): 2489-2495, 1995. PMID: 7593639.
- Defronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 58(4): 773-795, 2009. PMID: 19336687.
- DeFronzo RA. Lilly lecture 1987. The triumvirate: beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes 37(6): 667-687, 1988. PMID: 3289989.
- DeFronzo RA, Tobin JD and Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol Endocrinol Metab 237(3): E214-223, 1979. PMID: 382871.
- Dhillon S. Semaglutide: First Global Approval. Drugs 78(2): 275-284, 2018. PMID: 29363040.
- Dittrich HM. [History of the discovery of pancreatic diabetes by von Mering and Minkowski 1889. A historical overview on the occasion of the 100th anniversary]. Z Gesamte Inn Med 44(11): 335-340, 1989. PMID: 2669390.
- Edgerton DS, Lautz M, Scott M, Everett CA, Stettler KM, Neal DW, et al. Insulin's direct effects on the liver dominate the control of hepatic glucose production. J Clin Invest 116(2): 521-527, 2006. PMID: 16453026.
- Floyd JC, Jr., Fajans SS, Conn JW, Knopf RF and Rull J. Stimulation of insulin secretion by amino acids. J Clin Invest 45(9): 1487-1502, 1966. PMID: 5919350.
- Fred RG and Welsh N. The importance of RNA binding proteins in preproinsulin mRNA stability. Mol Cell Endocrinol 297(1-2): 28-33, 2009. PMID: 18621093.
- Fulgenzi G, Hong Z, Tomassoni-Ardori F, Barella LF, Becker J, Barrick C, et al. Novel metabolic role for BDNF in pancreatic β-cell insulin secretion. Nat Commun 11(1): 1950, 2020. PMID: 32327658.
- Gannon MC and Nuttall FQ. Amino acid ingestion and glucose metabolism--a review. IUBMB Life 62(9): 660-668, 2010. PMID: 20882645.
- Giddings SJ and Carnaghi L. Rat insulin II gene expression by extraplacental membranes. A non-pancreatic source for fetal insulin. J Biol Chem 264(16): 9462-9469, 1989. PMID: 2656699.
- Groop LC, Bonadonna RC, DelPrato S, Ratheiser K, Zyck K, Ferrannini E, et al. Glucose and free fatty acid metabolism in non-insulin-dependent diabetes mellitus. Evidence for multiple sites of insulin resistance. J Clin Invest 84(1): 205-213, 1989. PMID: 2661589.
- Guida C, McCulloch LJ, Godazgar M, Stephen SD, Baker C, Basco D, et al. Sitagliptin and Roux-en-Y gastric bypass modulate insulin secretion via regulation of intra-islet PYY. Diabetes Obes Metab 20(3): 571-581, 2018. PMID: 28892258.
- Guo XH. The value of short- and long-acting glucagon-like peptide-1 agonists in the management of type 2 diabetes mellitus: experience with exenatide. Curr Med Res Opin 32(1): 61-76, 2016. PMID: 26439329.
- Hatting M. Tavares CDJ, Sharabi K, Rines AK, and Puigserver P. Insulin regulation of gluconeogenesis. Ann N Y Acad Sci 1411(1): 21-35, 2018. PMID: 28868790.
- Hellman B. The frequency distribution of the number and volume of the islets Langerhans in man. I. Studies on non-diabetic adults. Acta Soc Med Ups 64: 432-460, 1959. PMID: 14400890.
- Henquin JC. The dual control of insulin secretion by glucose involves triggering and amplifying pathways in beta-cells. Diabetes Res Clin Pract 93 Suppl 1: S27-31, 2011. PMID: 21864748.
- Henquin JC. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49(11): 1751-1760, 2000. PMID: 11078440.
- Herman GA, Bergman A, Stevens C, Kotey P, Yi B, Zhao P, et al. Effect of single oral doses of sitagliptin, a dipeptidyl peptidase-4 inhibitor, on incretin and plasma glucose levels after an oral glucose tolerance test in patients with type 2 diabetes. J Clin Endocrinol Metab 91(11): 4612-9, 2006. PMID: 16912128.
- Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev 87(4): 1409-1439, 2007. PMID: 17928588.
- Holst JJ and Gromada J. Role of incretin hormones in the regulation of insulin secretion in diabetic and nondiabetic humans. Am J Physiol Endocrinol Metab 287(2): E199-206, 2004. PMID: 15271645.
- Hussain MA, Daniel PB and Habener JF. Glucagon stimulates expression of the inducible cAMP early repressor and suppresses insulin gene expression in pancreatic beta-cells. Diabetes 49(10): 1681-1690, 2000. PMID: 11016452.
- Jameson JL, De Kretser DM and DeGroot LJ (2010). Endocrinology: Adult and Pediatric. Philadelphia, Saunders / Elsevier.
- Jansson L and Hellerstrom C. Stimulation by glucose of the blood flow to the pancreatic islets of the rat. Diabetologia 25(1): 45-50, 1983. PMID: 6350083.
- Jones B, Bloom SR, Buenaventura T, Tomas A, and Rutter GA. Control of insulin secretion by GLP-1. Peptides 100: 75-84, 2018. PMID: 29412835.
- Jouvet N and Estall JL. The pancreas: Bandmaster of glucose homeostasis. Exp Cell Res 360(1): 19-23, 2017. PMID: 28351751.
- Kahn SE, D'Alessio DA, Schwartz MW, Fujimoto WY, Ensinck JW, Taborsky GJ, Jr., et al. Evidence of cosecretion of islet amyloid polypeptide and insulin by beta-cells. Diabetes 39(5): 634-638, 1990. PMID: 2185112.
- Karamanlis A, Chaikomin R, Doran S, Bellon M, Bartholomeusz FD, Wishart JM, et al. Effects of protein on glycemic and incretin responses and gastric emptying after oral glucose in healthy subjects. Am J Clin Nutr 86(5): 1364-1368, 2007. PMID: 17991647.
- Kashyap SR, Belfort R, Berria R, Suraamornkul S, Pratipranawatr T, Finlayson J, et al. Discordant effects of a chronic physiological increase in plasma FFA on insulin signaling in healthy subjects with or without a family history of type 2 diabetes. Am J Physiol Endocrinol Metab 287(3): E537-546, 2004. PMID: 15126243.
- Kock NG, Darle N, and Dotevall G. Inhibition of intestinal motility in man by glucagon given intraportally. Gastroenterology 53(1): 88-92, 1967. PMID: 6027233.
- Komatsu M, Takei M, Ishii H and Sato Y. Glucose-stimulated insulin secretion: A newer perspective. J Diabetes Investig 4(6): 511-516, 2013. PMID: 24843702.
- Lamont BJ, Li Y, Kwan E, Brown TJ, Gaisano H and Drucker DJ. Pancreatic GLP-1 receptor activation is sufficient for incretin control of glucose metabolism in mice. J Clin Invest 122(1): 388-402, 2012. PMID: 22182839.
- Lane MA. The cytological characters of the areas of langerhans. Am J Anat 7(3): 409-422, 1907.
- Lawrence MC, Bhatt HS and Easom RA. NFAT regulates insulin gene promoter activity in response to synergistic pathways induced by glucose and glucagon-like peptide-1. Diabetes 51(3): 691-698, 2002. PMID: 11872668.
- Le Roith D, Hendricks SA, Lesniak MA, Rishi S, Becker KL, Havrankova J, et al. Insulin in brain and other extrapancreatic tissues of vertebrates and nonvertebrates. Adv Metab Disord 10: 303-340, 1983. PMID: 6364717.
- Lebovitz HE and Melander A. Sulfonylureas and meglitinides: Insights into physiology and translational clinical utility. International Textbook of Diabetes Mellitus. DeFronzo RA, Ferrannini E, Zimmet P and Alberti G, John Wiley & Sons, Ltd. 1: 615, 2015.
- Lefèbvre PJ. Biosynthesis secretion and action of glucagon. In: International Textbook of Diabetes Mellitus. DeFronzo RA, Ferrannini E, Zimmet P and Alberti G, Eds. John Wiley & Sons, Ltd. 1: 136, 2015.
- Leibiger B, Moede T, Muhandiramlage TP, Kaiser D, Vaca Sanchez P, Leibiger IB, and Berggren PO. Glucagon regulates its own synthesis by autocrine signaling. Proc Natl Acad Sci U S A 109(51): 20925-20930, 2012. PMID: 23213228.
- Leto D and Saltiel AR. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat Rev Mol Cell Biol 13(6): 383-396, 2012. PMID: 22617471.
- Loubatieres-Mariani MM. [The discovery of hypoglycemic sulfonamides]. J Soc Biol 201(2): 121-125, 2007. PMID: 17978743.
- MacDonald PE and Rorsman P. The ins and outs of secretion from pancreatic beta-cells: control of single-vesicle exo- and endocytosis. Physiology (Bethesda) 22: 113-121, 2007. PMID: 17420302.
- Marchetti P, Bugliani M, De Tata V, Suleiman M, Marselli L. Pancreatic Beta Cell Identity in Humans and the Role of Type 2 Diabetes. Frontiers in Cell and Developmental Biology 5:55, 2017. PMID: 28589121.
- Masharani U and German MS. Pancreatic Hormones and Diabetes Mellitus. Greenspan’s Basic & Clinical Endocrinology, 9th Ed. Gardner DG and Shoback D. New York, NY, The McGraw-Hill Companies, 2011.
- Matschinsky FM and Wilson DF. The Central Role of Glucokinase in Glucose Homeostasis: A Perspective 50 Years After Demonstrating the Presence of the Enzyme in Islets of Langerhans. Front Physiol 10: 148, 2019. PMID: 30949058.
- Meglasson MD and Matschinsky FM. Pancreatic islet glucose metabolism and regulation of insulin secretion. Diabetes Metab Rev 2(3-4): 163-214, 1986. PMID: 2943567.
- Meier JJ, Nauck MA, Kranz D, Holst JJ, Deacon CF, Gaeckler D, et al. Secretion, degradation, and elimination of glucagon-like peptide 1 and gastric inhibitory polypeptide in patients with chronic renal insufficiency and healthy control subjects. Diabetes 53(3): 654-662, 2004. PMID: 14988249.
- Morisset J. Somatastatin. Pancreapedia: Exocrine Pancreas Knowledge Base, 2015. DOI: 10.3998/panc.2015.43.
- Morton GJ and Schwartz MW. Leptin and the central nervous system control of glucose metabolism. Physiol Rev 91(2): 389-411, 2011. PMID: 21527729.
- Murakami T, Fujita T, Taguchi T, Nonaka Y and Orita K. The blood vascular bed of the human pancreas, with special reference to the insulo-acinar portal system. Scanning electron microscopy of corrosion casts. Arch Histol Cytol 55(4): 381-395, 1992. PMID: 1482603.
- Muscelli E, Mari A, Natali A, Astiarraga BD, Camastra S, Frascerra S, et al. Impact of incretin hormones on beta-cell function in subjects with normal or impaired glucose tolerance. Am J Physiol Endocrinol Metab 291(6): E1144-1150, 2006. PMID: 16478775.
- Natali A, Del Prato S and Mari A. Normal β-cell function. International Textbook of Diabetes Mellitus. DeFronzo RA, Ferrannini E, Zimmet P and Alberti G, John Wiley & Sons, Ltd. 1: 108, 2015.
- Nuttall FQ, Gannon MC, Wald JL and Ahmed M. Plasma glucose and insulin profiles in normal subjects ingesting diets of varying carbohydrate, fat, and protein content. J Am Coll Nutr 4(4): 437-450, 1985. PMID: 3900180.
- O'Rahilly SO, Hosker JP, Rudenski AS, Matthews DR, Burnett MA and Turner RC. The glucose stimulus-response curve of the beta-cell in physically trained humans, assessed by hyperglycemic clamps. Metabolism 37(10): 919-923, 1988. PMID: 3050363.
- Ohtsubo K, Takamatsu S, Minowa MT, Yoshida A, Takeuchi M and Marth JD. Dietary and genetic control of glucose transporter 2 glycosylation promotes insulin secretion in suppressing diabetes. Cell 123(7): 1307-1321, 2005. PMID: 16377570.
- Philippe J. Somatostatin inhibits insulin-gene expression through a posttranscriptional mechanism in a hamster islet cell line. Diabetes 42(2): 244-249, 1993. PMID: 8093878.
- Pisania A, Weir GC, O'Neil JJ, Omer A, Tchipashvili V, Lei J, et al. Quantitative analysis of cell composition and purity of human pancreatic islet preparations. Lab Invest 90(11): 1661-1675, 2010. PMID: 20697378.
- Poitout V, Stein R and Rhodes CJ. Insulin gene expression and biosynthesis. In International Textbook of Diabetes Mellitus. DeFronzo RA, Ferrannini E, Zimmet P and Alberti G, eds, John Wiley & Sons, Ltd. 1: 82, 2015.
- Prentki M and Madiraju SR. Glycerolipid/free fatty acid cycle and islet beta-cell function in health, obesity and diabetes. Mol Cell Endocrinol 353(1-2): 88-100, 2012. PMID: 22108437.
- Prentki M, Matschinsky FM and Madiraju SR. Metabolic signaling in fuel-induced insulin secretion. Cell Metab 18(2): 162-185, 2013. PMID: 23791483.
- Quesada I, Nadal A and Soria B. Different effects of tolbutamide and diazoxide in alpha, beta-, and delta-cells within intact islets of Langerhans. Diabetes 48(12): 2390-2397, 1999. PMID: 10580428.
- Reaven G and Dray J. Effect of chlorpropamide on serum glucose and immunoreactive insulin concentrations in patients with maturity-onset diabetes mellitus. Diabetes 16(7): 487-492, 1967. PMID: 6028757.
- Röder PV, Wu B, Liu Y, and Han W. Pancreatic regulation of glucose homeostasis. Exp Mol Med 48(3): e219, 2016. PMID: 26964835.
- Rorsman P and Renstrom E. Insulin granule dynamics in pancreatic beta cells. Diabetologia 46(8): 1029-1045, 2003. PMID: 12879249.
- Rubenstein AH, Clark JL, Melani F and Steiner DF. Secretion of Proinsulin C-Peptide by Pancreatic [beta] Cells and its Circulation in Blood. Nature 224(5220): 697-699, 1969.
- Rudenski AS, Hosker JP, Burnett MA, Matthews DR and Turner RC. The beta cell glucose stimulus-response curve in normal humans assessed by insulin and C-peptide secretion rates. Metabolism 37(6): 526-534, 1988. PMID: 3287091.
- Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA and Butler PC. beta-cell mass and turnover in humans: effects of obesity and aging. Diabetes Care 36(1): 111-117, 2013. PMID: 22875233.
- Sanger F. Chemistry of insulin; determination of the structure of insulin opens the way to greater understanding of life processes. Science 129(3359): 1340-1344, 1959. PMID: 13658959.
- Schernthaner G, and Schernthaner GH. The right place for metformin today. Diabetes Res Clin Pract 159: 107946, 2020. PMID: 31778746.
- Schmid-Antomarchi H, de Weille J, Fosset M and Lazdunski M. The antidiabetic sulfonylurea glibenclamide is a potent blocker of the ATP-modulated K+ channel in insulin secreting cells. Biochem Biophys Res Commun 146(1): 21-25, 1987. PMID: 2440431.
- Schmid-Antomarchi H, De Weille J, Fosset M and Lazdunski M. The receptor for antidiabetic sulfonylureas controls the activity of the ATP-modulated K+ channel in insulin-secreting cells. J Biol Chem 262(33): 15840-15844, 1987. PMID: 2445740.
- Seino S, Shibasaki T and Minami K. β-Cell biology of insulin secretion. International Textbook of Diabetes Mellitus. DeFronzo RA, Ferrannini E, Zimmet P and Alberti G, John Wiley & Sons, Ltd. 1: 96, 2015.
- Şeker-Yılmaz B, Kör D, Bulut FD, Yüksel B, Karabay-Bayazıt A, Topaloğlu AK, et al. Impaired glucose tolerance in Fanconi-Bickel syndrome: Eight patients with two novel mutations. Turk J Pediatr 59 (4):434-441. 2017. PMID: 29624224.
- Shiratori K, Lee KY, Chang TM, Jo YH, Coy DH and Chey WY. Role of pancreatic polypeptide in the regulation of pancreatic exocrine secretion in dogs. Am J Physiol 255(5 Pt 1): G535-541, 1988. PMID: 3189545.
- Sindelar DK, Chu CA, Venson P, Donahue EP, Neal DW and Cherrington AD. Basal hepatic glucose production is regulated by the portal vein insulin concentration. Diabetes 47(4): 523-529, 1998. PMID: 9568682.
- Smith KM, Olson DC, Hirose R and Hanahan D. Pancreatic gene expression in rare cells of thymic medulla: evidence for functional contribution to T cell tolerance. Int Immunol 9(9): 1355-1365, 1997. PMID: 9310839.
- Smith PA, Sakura H, Coles B, Gummerson N, Proks P and Ashcroft FM. Electrogenic arginine transport mediates stimulus-secretion coupling in mouse pancreatic beta-cells. J Physiol 499 ( Pt 3): 625-635, 1997. PMID: 9130159.
- Stefan Y, Orci L, Malaisse-Lagae F, Perrelet A, Patel Y and Unger RH. Quantitation of endocrine cell content in the pancreas of nondiabetic and diabetic humans. Diabetes 31(8 Pt 1): 694-700, 1982. PMID: 6131002.
- Stein DT, Stevenson BE, Chester MW, Basit M, Daniels MB, Turley SD, et al. The insulinotropic potency of fatty acids is influenced profoundly by their chain length and degree of saturation. J Clin Invest 100(2): 398-403, 1997. PMID: 9218517.
- Stumvoll M, Meyer C, Mitrakou A, Nadkarni V and Gerich JE. Renal glucose production and utilization: new aspects in humans. Diabetologia 40(7): 749-757, 1997. PMID: 9243094.
- Thompson R, Christie D and Hindmarsh PC. The role for insulin analogues in diabetes care. Current Pediatrics 16(2): 117-122, 2006.
- Thorens B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia 58(2): 221-232, 2015. PMID: 25421524.
- Thorens B and Mueckler M. Glucose transporters in the 21st Century. Am J Physiol Endocrinol Metab 298(2): E141-145, 2010. PMID: 20009031.
- Thorens B, Sarkar HK, Kaback HR and Lodish HF. Cloning and functional expression in bacteria of a novel glucose transporter present in liver, intestine, kidney, and beta-pancreatic islet cells. Cell 55(2): 281-290, 1988. PMID: 3048704.
- Tong J, Prigeon RL, Davis HW, Bidlingmaier M, Kahn SE, Cummings DE, et al. Ghrelin suppresses glucose-stimulated insulin secretion and deteriorates glucose tolerance in healthy humans. Diabetes 59(9): 2145-2151, 2010. PMID: 20584998.
- Uchizono Y, Alarcon C, Wicksteed BL, Marsh BJ and Rhodes CJ. The balance between proinsulin biosynthesis and insulin secretion: where can imbalance lead? Diabetes Obes Metab 9 Suppl 2: 56-66, 2007. PMID: 17919179.
- Wang X, Misawa R, Zielinski MC, Cowen P, Jo J, Periwal V, et al. Regional differences in islet distribution in the human pancreas--preferential beta-cell loss in the head region in patients with type 2 diabetes. PLoS One 8(6): e67454, 2013. PMID: 23826303.
- Weiss R, Santoro N, Giannini G, Galderisi A, Umano GR, Caprio S. Prediabetes in youths: mechanisms and biomarkers. The Lancet Child & Adolescent Health 1(3): 240-248, 2017.
- Wendt A and Eliasson L. Pancreatic α-cells - The unsung heroes in islet function. Semin Cell Dev Biol 103: 41-50, 2020. PMID: 31983511.
- Williams JA. GLP-1. Pancreapedia: Exocrine Pancreas Knowledge Base, 2014. DOI: 10.3998/panc.2014.4
- Williams JA. Pancreatic Polypeptide. Pancreapedia: Exocrine Pancreas Knowledge Base, 2014. DOI: 10.3998/panc.2014.7
- Williams JA and Goldfine ID. The insulin-pancreatic acinar axis. Diabetes 34(10): 980-986, 1985. PMID: 2412919.
- Williams JA, Sankaran H, Korc M and Goldfine ID. Receptors for cholecystokinin and insulin in isolated pancreatic acini: hormonal control of secretion and metabolism. Fed Proc 40(10): 2497-2502, 1981. PMID: 6266879.
- Winzell MS and Ahren B. G-protein-coupled receptors and islet function-implications for treatmt of type 2 diabetes. Pharmacol Ther 116(3): 437-448, 2007. PMID: 17900700.
- Yu Q, Shuai QH, Ahooghalandari P, Gylfe E, and Tengholm A. Glucose controls glucagon secretion by directly modulating cAMP in alpha cells. Diabetologia 62(7): 1212-1224, 2019. PMID: 30953108.
- Zawalich WS and Zawalich KC. Species differences in the induction of time-dependent potentiation of insulin secretion. Endocrinology 137(5): 1664-1669, 1996. PMID: 8612499.