
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2021.08
| Attachment | Size |
|---|---|
| 520.03 KB |
I. Physiology of Pancreatic Ductal Cells
The main function of pancreatic ductal cells is to secrete a HCO3- rich, isotonic fluid that washes out the inactive form of digestive enzymes from the ductal system, as well as provide pH conditions that are essential for normal pancreatic function. The rate of HCO3- secretion is influenced by several factors (such as secretory rate, species, or the location of the cell in the ductal tree) and with stimulation, HCO3- can reach up to 140 mM. This means a significant concentration difference exists between the outside and inside of the ductal cell, which poses a physiologic challenge to duct cell homeostasis. The high level of HCO3- secretion is achieved through the coordinated action of ion transporters and channels, in which the Cl-/HCO3- exchanger and the cystic fibrosis transmembrane conductance regulator (CFTR) Cl- channel play a central role.
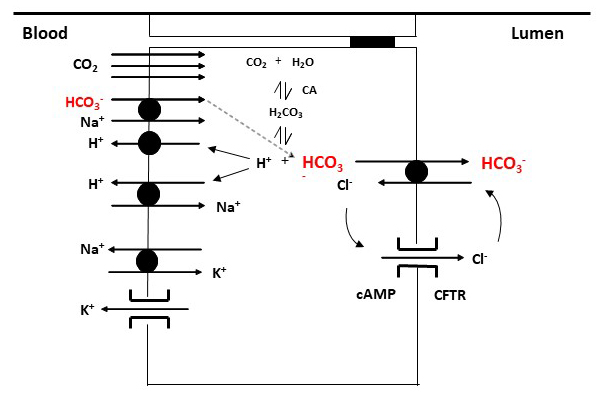
Ion transporters and channels on ductal cells are differentially expressed on the luminal and basolateral membranes, resulting in functional polarization of the ductal cell (Figure 1) (21). The major ion transporters on the duct cell basolateral membrane include the Na+/H+ exchanger (NHE), the Na+/HCO3- cotransporter (NBC), the Na+/K+ ATPase and various types of K+ channels (59). The electroneutral, NHE1 isoform can be found on the basolateral membrane of ductal cells and acts as a proton extruder with a 1 Na+:1 H+ stoichiometry (46). In addition to playing an important role in the regulation of intracellular pH, NHE1 promotes the formation of HCO3- from carbonic acid, by removing excess H+ from the cell. The NHE3 isoform is located on the luminal membrane of ductal cells and, unlike NHE1, is involved in HCO3- salvage (27, 35). HCO3- also enters the cell directly through NBC. The electrogenic NBC isoform, NBCe1B has been identified on the basolateral membrane of ductal cells, with 1 Na+/2 HCO3- stoichiometry (48, 55). Through this transporter, more HCO3- enters the cell than is formed during the dissociation of carbonic acid. The electroneutral form of NBC, the NBCn1 or NBC3, has been shown to be active on the luminal membrane of ductal cells, where it plays role in HCO3- salvage (43). In addition, Na+/K+ ATPase, together with basolateral K+ channels, maintains a negative membrane potential, using the energy from the hydrolysis of ATP, which provides a driving force for anion secretion across the luminal membrane.
There are two major transporters on the luminal side of the ductal cells; the CFTR chloride channel and the Cl-/HCO3- exchanger. Several mechanisms have been proposed to explain how ductal cells can secrete up to five-times as much HCO3- as is present in the cytosol (51, 52). The most generally accepted view is that HCO3- secretion occurs through both the Cl-/HCO3- exchanger and the CFTR channel (29, 42). Among the Cl-/HCO3- exchangers, the Slc26a6 (PAT1) and the Slc26a3 (DRA) isoforms are present on pancreatic ductal cells (15, 24, 54). PAT1 and DRA have different Cl- : HCO3- stoichiometry (2:1 for DRA and 1:2 for PAT1) and are expressed in different parts of the ductal tree. In the initial stage of HCO3- secretion, HCO3- is secreted into the lumen through the Cl-/HCO3- exchanger in exchange for Cl- that is returned to the lumen through the CFTR Cl- channel. Luminal concentration of HCO3- and Cl- follow a reciprocal pattern that are stimulation dependent. With increased secretory volume the concentration of HCO3- increases in the lumen and Cl- decreases. At the same time, the WNK1-OSR1/SPAK signalling pathway is activated, which increases the permeability of the CFTR channel to HCO3-, which allow HCO3- concentration to reach 140 mM HCO3 in the lumen (42).
Figure 1. Major ion transporters of pancreatic ductal cells. HCO3- enters the ductal cell directly via the basolateral Na+/HCO3- cotransporter (NBC). In addition, carbonic anhydrase (CA) is involved in the intracellular accumulation of HCO3- by catalyzing the formation of HCO3- and H+ from carbonic acid. The resulting H+ leaves the cell via the H+ pump or Na+/H+ exchanger (NHE). HCO3- is secreted into the lumen through the Cl-/HCO3- exchangers and the cystic fibrosis transmembrane conductance regulator (CFTR) Cl- channel, respectively.
II. Pathophysiology of Pancreatic Duct Cells
Normal electrolyte secretion is regained to maintain pancreatic exocrine cell homeostasis and solubilization of secretory protein; impairment of pancreatic fluid and electrolyte secretion contributes to tissue destruction in diseases such as Cystic Fibrosis (11, 12, 49, 53). An important consequence of impaired HCO3- secretion is an acidic pancreatic juice (less than 6.5). This increases mucus viscosity, and decreases the solubility of secreted digestive enzymes, which predisposes to the formation of mucin/protein plugs and eventually cysts within the ductal tree. A more acidic pH may also induce premature activation of digestive enzymes within the ductal tree, leading to the development of pancreatitis. Therefore, intensive research is underway to develop drug molecules capable of restoring the function of transporters, especially in CF.
A. Cystic fibrosis
CF is the most common, life-limiting, inherited disease in Caucasian populations (1 in 3,500 new-borns in Europe) (47, 53). Over 2000 CF causing mutations have been identified in the cftr gene (http://www.genet.sickkids.on.ca/), although to date, only ~ 360 of these variants have been fully annotated (https://cftr2.org/). However, 70-90% of CF individuals harbour the F508del mutation on at least one allele (10), which results in misfolding and incorrect processing of CFTR to the apical membrane. For those mutations that have been studied in detail, the genetic alteration leads to a variety of functional defects in the CFTR protein. These functional defects have been grouped into 6 classes (57). Class 1-3 cause severe CFTR dysfunction, while Class 4-6 produce less severe effects on CFTR and, in general, the mutated protein retains some level of channel activity. In relation to pancreatic pathology, ~ 85 % of people with CF are born pancreatic insufficient (PI), which equates to a reduction in pancreatic function of more than 95 %. In these people, there is a very good correlation between pancreatic disease severity and the class of mutation (9, 13, 62) with ‘severe’ CF mutations, such as the most common Class 2 CF mutation, F508del, and the Class 3 gating mutant, G511D, strongly correlate with PI. For those with ‘milder’ mutations (some residual channel activity such as Class 4, R117H), pancreatic function is preserved (pancreatic sufficient, PS), albeit to differing levels. However, in general these PS individuals require less pancreatic enzyme replacement supplements, but can become PI with increasing age. Mutations that cause moderate deficiencies in CFTR activity (10-50% of normal function) can increase the risk of developing pancreatitis, alone or when combined with other risk factors such as alcohol.
As described in the introduction, CFTR plays a fundamental role in pancreatic ductal NaHCO3 and fluid secretion, where it regulates HCO3- secretion in two fundamentally different ways; firstly, as a direct exit pathway for HCO3- secretion and secondly as a regulator of SLC26A-mediated Cl-/HCO3- exchange (21). For the latter process, this involves physical interaction between CFTR and the SLC26A6 anion exchanger (24, 25, 28, 30), and loss of functional CFTR leads to loss of anion exchange activity. The exact mechanism underlying this complete loss of anion exchange activity is not fully understood, but studies from polarised cultures of CFPAC cells, a human CF pancreatic ductal cell line homozygous for F508del, showed that apical SLC26A Cl-/HCO3-exchange activity was absent, despite evidence for mRNA expression. Importantly, viral-mediated CFTR transduction of the CFPAC cells restored anion exchange activity, suggesting that CFTR may be required for the trafficking of SLC26A6 to the apical membrane. Furthermore, it is interesting that the anion exchanger is activated by a number of CFTR mutants that lack Cl− channel activity (30), and that this correlates with a good retention of pancreatic function in patients carrying those mutations (6). Taken together, these results strongly suggest that a functional CFTR at the apical plasma membrane is a prerequisite for SLC26A6-mediated anion exchange, and that mild CFTR mutations are likely to preserve Cl-/HCO3- exchange activity, although this needs formal demonstration.
Strategies for improving HCO3- secretion in the CF pancreas are limited because of the marked tissue destruction at birth in the majority of people with CF. However, the last decade has seen a major improvement in the treatment of the basic defect in CF, through the development of small molecule CFTR modulators (44). Clinically-approved drugs include the CFTR potentiator, Ivacaftor, which enhances CFTR open probability for a number of gating mutants, as well as CFTR correctors, which help restore processing CFTR trafficking mutants (including F508del-CFTR) to the apical membrane. These include the first generation corrector drug, Lumacaftor and as well more efficacious second-generation correctors, such as tezacaftor and elexacaftor. These drugs are either given alone, or in combination (double and triple combinations), depending on the functional defects(s), and have produced substantial improvements in lung function, number of hospitalisations and exacerbations, as well as BMI (17, 37). Since lung dysfunction is the major cause of morbidity and mortality in people with CF, median survival rates are predicted to significantly improve. However, there are only a few studies that have directly assessed if these CFTR modulators also improve pancreatic function (36). For example, pancreatic function measurements in young children with CF taking Ivacaftor over 24 weeks, showed a significant restoration of enzyme-secreting capacity (increased faecal elastase-1 levels), and by inference, pancreatic tissue regeneration, which is an extremely exciting finding (8), that warrants further research. A more recent, but limited study, also provided evidence that Ivacaftor restored some pancreatic function in an adult with CF (http://dx.doi.org/10.3390/).
B. Acute pancreatitis
Acute pancreatitis (AP) is a sudden inflammation of the pancreas, which in most cases is mild, but in approximately 20% of patients, a life-threatening, severe form can develop in which the mortality rate can reach up to 20-40% (4). Large individual differences can be observed in the development and course of the disease, in which the disturbed balance of protective and damaging factors presumably plays a significant role (3). Pancreatic ductal cells are considered as protective mechanism in the pancreas by the secretion of a HCO3--rich, isotonic fluid. The two main etiological factors in the development of AP are gallstone obstruction and excessive alcohol consumption. Both aetiologies associated with marked changes in HCO3- and fluid secretion based on in vitro intracellular pH and fluid transport studies from isolated microdissected ducts (16, 20, 31, 34, 58, 60). At low concentrations, both agents increased HCO3- secretion, a response that required CFTR and Cl-/HCO3- exchange activity (18, 58, 60). However, higher levels of these agents led to a severe inhibition of CFTR-dependent HCO3‑ secretion, which was due to profound mitochondrial damage and a consequent reduction in intracellular ATP levels (20, 34) (Table 1). These studies were the first to suggest that ductal HCO3‑ secretion could play a protective role against these noxious agents.
Table 1. The effects of bile, ethanol and trypsin on pancreatic ductal HCO3- secretion.

Bile acids
Under normal conditions, bile cannot enter the pancreatic ductal system; however, in the case of gallstone obstruction it may regurgitate into the pancreas through the common bile duct. Chenodeoxycholate (CDCA) is the most abundant hydrophobic, unconjugated bile acid in human bile. Using isolated guinea pig pancreatic ducts, it has been shown that luminal administration of CDCA at low concentrations (0.1 mM), significantly increases ductal HCO3- secretion, in which CDCA-induced intracellular Ca2+ oscillation play an important role (60). The major source of CDCA-induced Ca2+ is the endoplasmic reticulum (ER), from which Ca2+ is released via an IP3-mediated pathway. The released Ca2+ activates large-conductance Ca2+-activated K+ channels (BKCa) on the luminal membrane of ductal cells, and opening of these channels hyperpolarizes the cell membrane, thereby enhancing the electrochemical driving force for anion secretion through the luminal membrane (58). This stimulatory effect of CDCA has also been demonstrated in the CFPAC-1 cell line and has been shown to be highly dependent on CFTR expression.(18) In contrast, high concentration of this bile acid (1 mM) causes a toxic increase in Ca2+ and strongly inhibits HCO3- secretion (60). The inhibitory effect of high concentrations of bile acids presumably results from the fact that at this concentration, CDCA damages mitochondria, resulting in ATP depletion and ultimately apoptosis (32-34, 60). This dual effect of bile acid is thought to be important in the pathogenesis of AP. In the early stages of the disease, when bile acids reach ductal cells at low concentrations, the increased fluid secretion try to wash out the toxic bile acids from the ductal tree to avoid pancreatic damage. If the concentration of bile acids further increases, it energetically destabilizes the cell, inhibits the function of ion transporters and induces apoptosis. Bile acids reach the acini where they induce inflammatory processes. Interestingly, the toxic effect of bile acids highly depends on their hydrophobicity. The hydrophilic bile acid, ursodeoxycholic acid, the 7-alpha enantiomer of CDCA, is able to counteract the cell-damaging effects of high-dose of CDCA through stabilization of the mitochondrial membrane that raises the possibility of therapeutic use of this bile acid (22, 56).
Ethanol
EtOH also dose-dependently affect ductal HCO3- secretion. Low concentration of EtOH (0.3-30 mM) enhances both basal and secretin-stimulated ductal fluid secretion from intra-interlobular ducts isolated from guinea pigs, in which cAMP activation and Ca2+ release play a key role (61). The stimulatory effect of low concentrations of EtOH, has also been demonstrated in the Capan-1 cell line and has been shown to be dependent on Ca2+ release from the ER via an IP3-mediated pathway (31). In contrast, high concentrations of EtOH (100 mM) strongly inhibited the rate of HCO3- secretion and the activity of CFTR (31, 61). The inhibitory effect of EtOH is presumably mediated by fatty acids (FAs) and fatty acid ethyl esters (FAEEs) formed during the non-oxidative metabolism of EtOH. Similar to the effect of bile acids, EtOH and its non-oxidative metabolites induce toxic Ca2+ signalling in ductal cells by completely depleting ER stores and promoting extracellular Ca2+ uptake into cells. Persistently elevated Ca2+ causes mitochondrial Ca2+ overload, resulting in decreased mitochondrial membrane potential and ATP production. Chelation of Ca2+ abolished the inhibitory effect of EtOH and fatty acid on HCO3- secretion, suggesting that the inhibitory effect of high dose of these agents is mediated by toxic intracellular Ca2+ (31). Moreover, EtOH and its metabolites profoundly inhibit CFTR function on the ductal cells which can be prevented by the supplementation of intracellular ATP (ATPi), indicating that the inhibitory effect of EtOH on CFTR is mediated by ATPi depletion. Since CFTR works in close coordination with the Cl-/HCO3- exchanger, improper CFTR function may contribute to decreased fluid and HCO3- secretion and thus to the pathogenesis of AP. Consequently, maintenance of ATPi may represent a therapeutic option in the treatment of the disease (20, 31). Decreased expression of CFTR has been also observed on the luminal membrane of human pancreatic ductal cells in alcohol-induced acute and chronic pancreatitis, and in response to FAs and FAEEs in which the accelerated turnover and decreased biosynthesis of the channel play role. The importance of CFTR in the alcohol-induced pancreatic damage has been further confirmed in CFTR knock out mice, where the absence of CFTR caused much more severe pancreatitis (31).
Trypsin
One of the most important roles of ductal HCO3- secretion is to prevent intraductal activation of trypsinogen. Although there is no significant trypsin in the lumen under physiological conditions, it may leak from the acinar cells in the early stages of pancreatitis. By binding to PAR-2 receptors on the luminal membrane of ductal cells, trypsin or trypsin activating peptide (PAR-2-AP) increases [Ca2+]i levels and inhibits Cl-/HCO3- exchange and CFTR function, resulting in lower luminal pH (41). Low luminal pH favours premature activation of trypsinogen, which will activate additional trypsinogen molecules. This process leads to a vicious cycle in which more trypsin is formed, the more trypsinogen will be activated, resulting in even more inhibition of the activity of the luminal transporters (41). The importance of PAR-2 activation in the pathobiology of pancreatitis has been also demonstrated using PAR-2 knock out mice, in which luminal administration of either trypsin or PAR-2-AP had significantly lower effect on both [Ca2+]i and pHi.
III. Therapeutic Perspectives and Clinical Significance
The CFTR chloride channel is clearly the most investigated and most utilized ductal channel that has been targeted for therapy (23, 50). In the first decades after the discovery of the CFTR gene, only symptomatic therapy was available. Difficulties over the years have been caused by the heterogeneity of CFTR gene mutations. Therefore, it is almost needless to say, that CF is typically a disease where personalized therapy needs to be considered. However, one approach that would be potentially be suitable for all people with CF, is gene therapy. The first randomized clinical trial, of a non-viral-based gene therapy for CF, was performed by E. Alton et al. They found that a 12-month-treatment by pGM169/G67A gene therapy formulation improved lung function among the CF patients (2). Although the results were very promising, no further trials have taken place since the original observation, although pre-clinical development of a viral-mediated gene therapy treatment for CF is underway (https://www.cff.org/Trials/Pipeline/details/10160/Spirovant-Sciences). However, in the last decade the orally bioavailable correctors, potentiators and suppressors of CFTR gene mutations have become available for treatment (39).
CFTR-directed therapies may also be useful for the treatment of pancreatitis, since recent animal studies have suggested that strategies that help maintain levels of HCO3‑ secretion would limit the extent of pathology induced by bile and alcohol (31, 34, 40). Furthermore, the effects of ethanol and ethanol metabolites on CFTR are consistent with reduced biogenesis, accelerated plasma membrane turnover, as well as channel inhibition (31). Thus, restoring cell surface expression and activity of CFTR could partially alleviate the ethanol-induced damage. This potentially could be through use of CFTR correctors (Lumacaftor, Tezacaftor), as well as potentiators (Ivacaftor) to improve channel activity. We have recently found that Ivacaftor (VX-770) and Lumacaftor (VX-809) restore CFTR expression defect associated with alcohol in pancreatic ductal cells, suggesting that Orkambi® may serve as a therapeutic option in acute, recurrent or chronic pancreatitis (14). Akshintala et al. recently showed that CFTR modulators, alone or in combination, reduced the risk of recurrent acute pancreatitis within a 3-year-follow up therapy in adult CF patients (1). A 24-year-old CF patient with R117H/7 T/F508del mutations with recurrent acute pancreatitis were reported pancreatitis free during ivacaftor therapy (19). Carrion et al. also found a reduced frequency and recurrence rate of pancreatitis in patients with CF during Ivacaftor therapy (5). Both the basic research results and the pancreatitis-free periods achieved in CF patients suggest that the drugs used for treating CF patients should be tested in randomized clinical trials in non-CF patients with recurrent pancreatitis as well.
Another possible way to compensate for defective CFTR would be to target alternative ion channels, such as the calcium-activated Cl channel, TMEM16A (7, 45), or transporters such as the SLC26 family members (A3, A6 or A9), or short-circuiting their regulation by CFTR and rebalancing exocrine homeostasis. It has been shown that variants (SNPs) in the SLC26A9 anion transporter influence disease severity in the CF lungs and intestinal tract, and therefore act as gene modifiers. Importantly, a recent study has suggested that SNPs in SLC26A9 also influence the degree of pancreatic insufficiency (38). Furthermore, variants of SLC26A9 also influence the extent of CF-related diabetes, which may be due to beneficial effects of restoring ductal bicarbonate secretion on endocrine (islet) function in CF (26). This opens up the possibility of targeting this anion transporter as a potential therapeutic target to slow the progression of exocrine dysfunction in CF. One important advantage of this ‘alternate non-CFTR approach’ is that it would benefit all CF patients regardless of genotype.
IV. Summary
One of the most important functions of ductal cells is their ability to neutralize acidic pH within the pancreas and duodenum. This ability of ductal cells is due to the secretion of a HCO3- rich fluid, which results from the coordinated action of ductal ion channels and transporters. Impairment of transport processes can result in a decrease in both the volume and pH of pancreatic fluid, which can predispose to inflammatory processes and consequently the development of various diseases. In the case of CF, it is well known that the inadequate function of the CFTR channel underlies the disease, however, it is only recently that research has shed light on the pathological role of ductal cells in pancreatitis. Various etiological factors such as bile acids and EtOH are now known to impair ductal HCO3- and fluid secretion, which are likely to play an important role in initiating pancreatitis by creating an unfavorable pH environment. Consequently, drugs that enhance the function of ion transporters may be of great importance not only in CF therapy, but also in the treatment of pancreatitis.
V. References.
- Akshintala VS, Kamal A, Faghih M, Cutting GR, Cebotaru L, West NE, Jennings MT, Dezube R, Whitcomb DC, Lechtzin N, Merlo CA, and Singh VK. Cystic fibrosis transmembrane conductance regulator modulators reduce the risk of recurrent acute pancreatitis among adult patients with pancreas sufficient cystic fibrosis. Pancreatology 19: 1023-1026, 2019. PMID: 31611131.
- Alton E, Armstrong DK, Ashby D, Bayfield KJ, Bilton D, Bloomfield EV, Boyd AC, Brand J, Buchan R, Calcedo R, Carvelli P, Chan M, Cheng SH, Collie DDS, Cunningham S, Davidson HE, Davies G, Davies JC, Davies LA, Dewar MH, Doherty A, Donovan J, Dwyer NS, Elgmati HI, Featherstone RF, Gavino J, Gea-Sorli S, Geddes DM, Gibson JSR, Gill DR, Greening AP, Griesenbach U, Hansell DM, Harman K, Higgins TE, Hodges SL, Hyde SC, Hyndman L, Innes JA, Jacob J, Jones N, Keogh BF, Limberis MP, Lloyd-Evans P, Maclean AW, Manvell MC, McCormick D, McGovern M, McLachlan G, Meng C, Montero MA, Milligan H, Moyce LJ, Murray GD, Nicholson AG, Osadolor T, Parra-Leiton J, Porteous DJ, Pringle IA, Punch EK, Pytel KM, Quittner AL, Rivellini G, Saunders CJ, Scheule RK, Sheard S, Simmonds NJ, Smith K, Smith SN, Soussi N, Soussi S, Spearing EJ, Stevenson BJ, Sumner-Jones SG, Turkkila M, Ureta RP, Waller MD, Wasowicz MY, Wilson JM, Wolstenholme-Hogg P, and Consortium UKCFGT. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir Med 3: 684-691, 2015. PMID: 26149841.
- Barreto SG, Habtezion A, Gukovskaya A, Lugea A, Jeon C, Yadav D, Hegyi P, Venglovecz V, Sutton R, and Pandol SJ. Critical thresholds: key to unlocking the door to the prevention and specific treatments for acute pancreatitis. Gut 20: 194-203. 2020. PMID: 32973069.
- Boxhoorn L, Voermans RP, Bouwense SA, Bruno MJ, Verdonk RC, Boermeester MA, van Santvoort HC, and Besselink MG. Acute pancreatitis. Lancet 396: 726-734, 2020. PMID: 32891214.
- Carrion A, Borowitz DS, Freedman SD, Siracusa CM, Goralski JL, Hadjiliadis D, Srinivasan S, and Stokes DC. Reduction of Recurrence Risk of Pancreatitis in Cystic Fibrosis With Ivacaftor: Case Series. J Pediatr Gastroenterol Nutr 66: 451-454, 2018. PMID: 29045327.
- Choi JY, Muallem D, Kiselyov K, Lee MG, Thomas PJ, and Muallem S. Aberrant CFTR-dependent HCO3- transport in mutations associated with cystic fibrosis. Nature 410: 94-97, 2001. PMID: 11242048.
- Danahay H, and Gosling M. TMEM16A: An Alternative Approach to Restoring Airway Anion Secretion in Cystic Fibrosis? Int J Mol Sci 21: 2020. PMID: 32235608.
- Davies JC, Cunningham S, Harris WT, Lapey A, Regelmann WE, Sawicki GS, Southern KW, Robertson S, Green Y, Cooke J, Rosenfeld M, and Group KS. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2-5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med 4: 107-115, 2016. PMID: 26803277.
- De Boeck K, Weren M, Proesmans M, and Kerem E. Pancreatitis among patients with cystic fibrosis: correlation with pancreatic status and genotype. Pediatrics 115: e463-469, 2005. PMID: 15772171.
- De Boeck K, Zolin A, Cuppens H, Olesen HV, and Viviani L. The relative frequency of CFTR mutation classes in European patients with cystic fibrosis. J Cyst Fibros 13: 403-409, 2014. PMID: 24440181.
- Durie PR. Pancreatitis and mutations of the cystic fibrosis gene. N Engl J Med 339: 687-688, 1998. PMID: 9725928.
- Durie PR. The pathophysiology of the pancreatic defect in cystic fibrosis. Acta Paediatr Scand Suppl 363: 41-44, 1989. PMID: 2701923.
- Durno C, Corey M, Zielenski J, Tullis E, Tsui LC, and Durie P. Genotype and phenotype correlations in patients with cystic fibrosis and pancreatitis. Gastroenterology 123: 1857-1864, 2002. PMID: 12454843.
- Grassalkovich A MJ, Madácsy T, Venglovecz V, Hegyi P. VX-770 and VX-809 restore the alcohol-induced CFTR expression defect in pancreatic ductal cells. In: European Pancreatic Club. Bergen, Norway: Elsevier, 2019, p. S28.
- Greeley T, Shumaker H, Wang Z, Schweinfest CW, and Soleimani M. Downregulated in adenoma and putative anion transporter are regulated by CFTR in cultured pancreatic duct cells. Am J Physiol Gastrointest Liver Physiol 281: G1301-1308, 2001. PMID: 11668039.
- Hegyi P, Pandol S, Venglovecz V, and Rakonczay Z, Jr. The acinar-ductal tango in the pathogenesis of acute pancreatitis. Gut 60: 544-552, 2011. PMID: 20876773.
- Heijerman HGM, McKone EF, Downey DG, Van Braeckel E, Rowe SM, Tullis E, Mall MA, Welter JJ, Ramsey BW, McKee CM, Marigowda G, Moskowitz SM, Waltz D, Sosnay PR, Simard C, Ahluwalia N, Xuan F, Zhang Y, Taylor-Cousar JL, McCoy KS, and Group VXT. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet 394: 1940-1948, 2019. PMID: 31679946.
- Ignath I, Hegyi P, Venglovecz V, Szekely CA, Carr G, Hasegawa M, Inoue M, Takacs T, Argent BE, Gray MA, and Rakonczay Z, Jr. CFTR expression but not Cl- transport is involved in the stimulatory effect of bile acids on apical Cl-/HCO3- exchange activity in human pancreatic duct cells. Pancreas 38: 921-929, 2009. PMID: 19752774.
- Johns JD, and Rowe SM. The effect of CFTR modulators on a cystic fibrosis patient presenting with recurrent pancreatitis in the absence of respiratory symptoms: a case report. BMC Gastroenterol 19: 123, 2019. PMID: 31296159.
- Judak L, Hegyi P, Rakonczay Z, Jr., Maleth J, Gray MA, and Venglovecz V. Ethanol and its non-oxidative metabolites profoundly inhibit CFTR function in pancreatic epithelial cells which is prevented by ATP supplementation. Pflugers Arch 466: 549-562, 2014. PMID: 23948742.
- Lee MG, Kim Y, Jun I, Aoun J, and Muallem S. Molecular Mechanisms of Pancreatic Bicarbonate Secretion. Pancreapedia 2021. DOI: 10.3998/panc.2020.06.
- Katona M, Hegyi P, Kui B, Balla Z, Rakonczay Z, Jr., Razga Z, Tiszlavicz L, Maleth J, and Venglovecz V. A novel, protective role of ursodeoxycholate in bile-induced pancreatic ductal injury. Am J Physiol Gastrointest Liver Physiol 310: G193-204, 2016. PMID: 26608189.
- Kleizen B, Hunt JF, Callebaut I, Hwang TC, Sermet-Gaudelus I, Hafkemeyer S, and Sheppard DN. CFTR: New insights into structure and function and implications for modulation by small molecules. J Cyst Fibros 19 Suppl 1: S19-S24, 2020. PMID: 31759907.
- Ko SB, Shcheynikov N, Choi JY, Luo X, Ishibashi K, Thomas PJ, Kim JY, Kim KH, Lee MG, Naruse S, and Muallem S. A molecular mechanism for aberrant CFTR-dependent HCO(3)(-) transport in cystic fibrosis. EMBO J 21: 5662-5672, 2002. PMID: 12411484.
- Ko SB, Zeng W, Dorwart MR, Luo X, Kim KH, Millen L, Goto H, Naruse S, Soyombo A, Thomas PJ, and Muallem S. Gating of CFTR by the STAS domain of SLC26 transporters. Nat Cell Biol 6: 343-350, 2004. PMID: 15048129.
- Lam AN, Aksit MA, Vecchio-Pagan B, Shelton CA, Osorio DL, Anzmann AF, Goff LA, Whitcomb DC, Blackman SM, and Cutting GR. Increased expression of anion transporter SLC26A9 delays diabetes onset in cystic fibrosis. J Clin Invest 130: 272-286, 2020. PMID: 31581148.
- Lee MG, Ahn W, Choi JY, Luo X, Seo JT, Schultheis PJ, Shull GE, Kim KH, and Muallem S. Na(+)-dependent transporters mediate HCO(3)(-) salvage across the luminal membrane of the main pancreatic duct. J Clin Invest 105: 1651-1658, 2000. PMID: 10841524.
- Lee MG, Choi JY, Luo X, Strickland E, Thomas PJ, and Muallem S. Cystic fibrosis transmembrane conductance regulator regulates luminal Cl-/HCO3- exchange in mouse submandibular and pancreatic ducts. J Biol Chem 274: 14670-14677, 1999. PMID: 10329661.
- Lee MG, Ohana E, Park HW, Yang D, and Muallem S. Molecular mechanism of pancreatic and salivary gland fluid and HCO3 secretion. Physiol Rev 92: 39-74, 2012. PMID: 22298651.
- Lee MG, Wigley WC, Zeng W, Noel LE, Marino CR, Thomas PJ, and Muallem S. Regulation of Cl-/ HCO3- exchange by cystic fibrosis transmembrane conductance regulator expressed in NIH 3T3 and HEK 293 cells. J Biol Chem 274: 3414-3421, 1999. PMID: 9920885.
- Maleth J, Balazs A, Pallagi P, Balla Z, Kui B, Katona M, Judak L, Nemeth I, Kemeny LV, Rakonczay Z, Jr., Venglovecz V, Foldesi I, Peto Z, Somoracz A, Borka K, Perdomo D, Lukacs GL, Gray MA, Monterisi S, Zaccolo M, Sendler M, Mayerle J, Kuhn JP, Lerch MM, Sahin-Toth M, and Hegyi P. Alcohol disrupts levels and function of the cystic fibrosis transmembrane conductance regulator to promote development of pancreatitis. Gastroenterology 148: 427-439 e416, 2015. PMID: 25447846.
- Maleth J, Hegyi P, Rakonczay Z, Jr., and Venglovecz V. Breakdown of bioenergetics evoked by mitochondrial damage in acute pancreatitis: Mechanisms and consequences. Pancreatology 15: S18-22, 2015. PMID: 26162756.
- Maleth J, Rakonczay Z, Jr., Venglovecz V, Dolman NJ, and Hegyi P. Central role of mitochondrial injury in the pathogenesis of acute pancreatitis. Acta Physiol (Oxf) 207: 226-235, 2013. PMID: 23167280.
- Maleth J, Venglovecz V, Razga Z, Tiszlavicz L, Rakonczay Z, Jr., and Hegyi P. Non-conjugated chenodeoxycholate induces severe mitochondrial damage and inhibits bicarbonate transport in pancreatic duct cells. Gut 60: 136-138, 2011. PMID: 20732916.
- Marteau C, Silviani V, Ducroc R, Crotte C, and Gerolami A. Evidence for apical Na+/H+ exchanger in bovine main pancreatic duct. Dig Dis Sci 40: 2336-2340, 1995. PMID: 7587811.
- Megalaa R, Gopalareddy V, Champion E, and Goralski JL. Time for a gut check: Pancreatic sufficiency resulting from CFTR modulator use. Pediatr Pulmonol 54: E16-E18, 2019. PMID: 31066218.
- Middleton PG, Mall MA, Drevinek P, Lands LC, McKone EF, Polineni D, Ramsey BW, Taylor-Cousar JL, Tullis E, Vermeulen F, Marigowda G, McKee CM, Moskowitz SM, Nair N, Savage J, Simard C, Tian S, Waltz D, Xuan F, Rowe SM, Jain R, and Group VXS. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N Engl J Med 381: 1809-1819, 2019. PMID: 31697873.
- Miller MR, Soave D, Li W, Gong J, Pace RG, Boelle PY, Cutting GR, Drumm ML, Knowles MR, Sun L, Rommens JM, Accurso F, Durie PR, Corvol H, Levy H, Sontag MK, and Strug LJ. Variants in Solute Carrier SLC26A9 Modify Prenatal Exocrine Pancreatic Damage in Cystic Fibrosis. J Pediatr 166: 1152-1157 e1156, 2015. PMID: 25771386.
- Nichols DP, Donaldson SH, Frederick CA, Freedman SD, Gelfond D, Hoffman LR, Kelly A, Narkewicz MR, Pittman JE, Ratjen F, Sagel SD, Rosenfeld M, Schwarzenberg SJ, Singh PK, Solomon GM, Stalvey MS, Kirby S, VanDalfsen JM, Clancy JP, and Rowe SM. PROMISE: Working with the CF community to understand emerging clinical and research needs for those treated with highly effective CFTR modulator therapy. J Cyst Fibros 2021. PMID: 33619012.
- Pallagi P, Balla Z, Singh AK, Dosa S, Ivanyi B, Kukor Z, Toth A, Riederer B, Liu Y, Engelhardt R, Jarmay K, Szabo A, Janovszky A, Perides G, Venglovecz V, Maleth J, Wittmann T, Takacs T, Gray MA, Gacser A, Hegyi P, Seidler U, and Rakonczay Z, Jr. The role of pancreatic ductal secretion in protection against acute pancreatitis in mice*. Crit Care Med 42: e177-188, 2014. PMID: 24368347.
- Pallagi P, Venglovecz V, Rakonczay Z, Jr., Borka K, Korompay A, Ozsvari B, Judak L, Sahin-Toth M, Geisz A, Schnur A, Maleth J, Takacs T, Gray MA, Argent BE, Mayerle J, Lerch MM, Wittmann T, and Hegyi P. Trypsin reduces pancreatic ductal bicarbonate secretion by inhibiting CFTR Cl(-) channels and luminal anion exchangers. Gastroenterology 141: 2228-2239 e2226, 2011. PMID: 21893120.
- Park HW, Nam JH, Kim JY, Namkung W, Yoon JS, Lee JS, Kim KS, Venglovecz V, Gray MA, Kim KH, and Lee MG. Dynamic regulation of CFTR bicarbonate permeability by [Cl-]i and its role in pancreatic bicarbonate secretion. Gastroenterology 139: 620-631, 2010. PMID: 20398666.
- Park M, Ko SB, Choi JY, Muallem G, Thomas PJ, Pushkin A, Lee MS, Kim JY, Lee MG, Muallem S, and Kurtz I. The cystic fibrosis transmembrane conductance regulator interacts with and regulates the activity of the HCO3- salvage transporter human Na+-HCO3- cotransport isoform 3. J Biol Chem 277: 50503-50509, 2002. PMID: 12403779.
- Patel SD, Bono TR, Rowe SM, and Solomon GM. CFTR targeted therapies: recent advances in cystic fibrosis and possibilities in other diseases of the airways. Eur Respir Rev 29: 190068, 2020. PMID: 32554756.
- Quesada R, and Dutzler R. Alternative chloride transport pathways as pharmacological targets for the treatment of cystic fibrosis. J Cyst Fibros 19 Suppl 1: S37-S41, 2020. PMID: 31662238.
- Roussa E, Alper SL, and Thevenod F. Immunolocalization of anion exchanger AE2, Na(+)/H(+) exchangers NHE1 and NHE4, and vacuolar type H(+)-ATPase in rat pancreas. J Histochem Cytochem 49: 463-474, 2001. PMID: 11259449.
- Salvatore D, Buzzetti R, Baldo E, Forneris MP, Lucidi V, Manunza D, Marinelli I, Messore B, Neri AS, Raia V, Furnari ML, and Mastella G. An overview of international literature from cystic fibrosis registries. Part 3. Disease incidence, genotype/phenotype correlation, microbiology, pregnancy, clinical complications, lung transplantation, and miscellanea. J Cyst Fibros 10: 71-85, 2011. PMID: 21257352.
- Satoh H, Moriyama N, Hara C, Yamada H, Horita S, Kunimi M, Tsukamoto K, Iso ON, Inatomi J, Kawakami H, Kudo A, Endou H, Igarashi T, Goto A, Fujita T, and Seki G. Localization of Na+-HCO-3 cotransporter (NBC-1) variants in rat and human pancreas. Am J Physiol Cell Physiol 284: C729-737, 2003. PMID: 12444017.
- Scheele GA, Fukuoka SI, Kern HF, and Freedman SD. Pancreatic dysfunction in cystic fibrosis occurs as a result of impairments in luminal pH, apical trafficking of zymogen granule membranes, and solubilization of secretory enzymes. Pancreas 12: 1-9, 1996. PMID: 8927611.
- Schneider-Futschik EK. Beyond cystic fibrosis transmembrane conductance regulator therapy: a perspective on gene therapy and small molecule treatment for cystic fibrosis. Gene Ther 26: 354-362, 2019. PMID: 31300729.
- Sohma Y, Gray MA, Imai Y, and Argent BE. 150 mM HCO3(-)--how does the pancreas do it? Clues from computer modelling of the duct cell. JOP 2: 198-202, 2001. PMID: 11875259.
- Sohma Y, Gray MA, Imai Y, and Argent BE. HCO3- transport in a mathematical model of the pancreatic ductal epithelium. J Membr Biol 176: 77-100, 2000. PMID: 10882430.
- Southern KW, Munck A, Pollitt R, Travert G, Zanolla L, Dankert-Roelse J, Castellani C, and Group ECNSW. A survey of newborn screening for cystic fibrosis in Europe. J Cyst Fibros 6: 57-65, 2007. PMID: 16870510.
- Stewart AK, Yamamoto A, Nakakuki M, Kondo T, Alper SL, and Ishiguro H. Functional coupling of apical Cl-/HCO3- exchange with CFTR in stimulated HCO3- secretion by guinea pig interlobular pancreatic duct. Am J Physiol Gastrointest Liver Physiol 296: G1307-1317, 2009. PMID: 19342507.
- Thevenod F, Roussa E, Schmitt BM, and Romero MF. Cloning and immunolocalization of a rat pancreatic Na(+) bicarbonate cotransporter. Biochem Biophys Res Commun 264: 291-298, 1999. PMID: 10527880.
- Tsubakio K, Kiriyama K, Matsushima N, Taniguchi M, Shizusawa T, Katoh T, Manabe N, Yabu M, Kanayama Y, and Himeno S. Autoimmune pancreatitis successfully treated with ursodeoxycholic acid. Intern Med 41: 1142-1146, 2002. PMID: 12521203.
- Veit G, Avramescu RG, Chiang AN, Houck SA, Cai Z, Peters KW, Hong JS, Pollard HB, Guggino WB, Balch WE, Skach WR, Cutting GR, Frizzell RA, Sheppard DN, Cyr DM, Sorscher EJ, Brodsky JL, and Lukacs GL. From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol Biol Cell 27: 424-433, 2016. PMID: 26823392.
- Venglovecz V, Hegyi P, Rakonczay Z, Jr., Tiszlavicz L, Nardi A, Grunnet M, and Gray MA. Pathophysiological relevance of apical large-conductance Ca(2)+-activated potassium channels in pancreatic duct epithelial cells. Gut 60: 361-369, 2011. PMID: 20940280.
- Venglovecz V, Rakonczay Z, Jr., Gray MA, and Hegyi P. Potassium channels in pancreatic duct epithelial cells: their role, function and pathophysiological relevance. Pflugers Arch 467: 625-640, 2015. PMID: 25074489.
- Venglovecz V, Rakonczay Z, Jr., Ozsvari B, Takacs T, Lonovics J, Varro A, Gray MA, Argent BE, and Hegyi P. Effects of bile acids on pancreatic ductal bicarbonate secretion in guinea pig. Gut 57: 1102-1112, 2008. PMID: 18303091.
- Yamamoto A, Ishiguro H, Ko SB, Suzuki A, Wang Y, Hamada H, Mizuno N, Kitagawa M, Hayakawa T, and Naruse S. Ethanol induces fluid hypersecretion from guinea-pig pancreatic duct cells. J Physiol 551: 917-926, 2003. PMID: 12847207.
- Zielenski J. Genotype and phenotype in cystic fibrosis. Respiration 67: 117-133, 2000. PMID: 10773783.