
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2013.4
| Attachment | Size |
|---|---|
| 225.43 KB |
General Comments
Acute pancreatitis (AP) is a disorder with a sudden onset that can present in mild edematous and severe necrotizing forms (50). Mild AP is usually self-limiting and is characterized by minimal or no distant organ dysfunction and an uncomplicated recovery. In contrast, severe necrotizing AP usually involves organ failure (pulmonary insufficiency, renal failure, shock, etc.), and local complications (such as infected necrosis, abscess, or pseudocyst formation). These complications of severe AP contribute to its high mortality and morbidity rates.
Despite intensive research, our understanding of the pathophysiology of AP is far from complete. Because it is generally not possible to obtain pancreatic tissue during the early stages of the clinical disease, most of our knowledge-base comes from studies using in vivo animal models of AP (2, 11, 56, 60, 68). Nowadays, ex vivo models of AP are also becoming more widely used, especially for mechanistic studies. On the other hand, as AP is a systemic disease involving other organs besides the pancreas, the usefulness of ex vivo AP models is still limited and the preferred use of in vivo AP models in investigating AP is more than justified.
About 80% of human AP cases are related to either ethanol abuse or gall stone disease. However, the disease will develop in only a minority (£10%) of individuals who either harbor gall stones or consume alcohol and there are no animal models of AP that can be induced by these conditions alone. Conversely most of the agents commonly used to induce experimental AP in animals do not induce pancreatitis in humans.
Necrotizing clinical AP is usually characterized by large, patchy areas of hemorrhagic necrosis of pancreatic and peripancreatic tissues (28, 29, 45) and it is not characterized by diffuse, homogeneous injury. The necrosis of severe clinical pancreatitis usually develops within the first 4 days after the onset of symptoms in humans, whereas infection of the necrotic pancreas develops most frequently in the second and third weeks after the onset of symptoms (4). Actually, many of the deaths in necrotizing AP result from pancreatic infections, which are reported to occur in 30% to 70% of patients (5). The incidence of infection correlates with the extent of intra- and extrapancreatic tissue necrosis. Rodents with necrotizing AP seem to be much more resistant to infections than are humans and considerable pancreatic infection rates are only reported for the invasive rodent AP models (70).
With respect to treatment of the disease, researchers have often found that drugs which are beneficial in the treatment of rodent models of experimental pancreatitis fail to be effective in clinical trials. This may be because most treatment protocols in animals permit starting treatment before or very shortly after the induction of AP and, therefore, this does not resemble the clinical situation in which prophylactic therapy is only possible in cases of endoscopic retrograde cholangiopancreatography-induced AP.
It is likely that the rate of AP progression differs between experimental and clinical pancreatitis. Disease kinetics appears to depend on many parameters, including body mass and, while many patients with severe pancreatitis are also obese, most animal models of pancreatitis are based on the use of lean animals. At the time of emergency room presentation, which is usually 12-36 hours after the onset of symptoms, the clinical disease is usually already quite developed (11). In contrast, pronounced pathology in some models [i.e. choline-deficient ethionine-supplemented (CDE)-diet, L-arginine] requires many hours to develop.
Despite these various potentially important differences between clinical and experimental pancreatitis, we have come a long way since the first experimental AP model (retrograde injection of bile and olive oil into the pancreatic duct of a dog) was described by Claude Bernard in 1856 (6). Since then, numerous AP models have been developed. In the past, large animals such as dogs and cats were commonly used in AP studies, but nowadays most investigations are performed on small animals, usually rats and mice. The latter species has become increasingly used due, primarily, to the availability of genetically modified mouse strains. Although the use of larger animals may present fewer technical limitations related to the size of the subjects [such as surgical or therapeutic interventions including intravenous (i.v.) administration of drugs and fluids], rodents are also utilized more commonly because of financial, ethical and practical reasons. Furthermore, in-bred strains of rodents are better standardized and the utilization of larger numbers of animals per group can improve the statistical power of the experiments. In this regard, the use of in-bred strains of rodents may offer distinct advantages over clinical studies since, in the latter case, it is often difficult to recruit and monitor a sufficiently large and homogeneous population of patients with which to evaluate the effects of therapeutic interventions.
Aims
The main aims of this review are to provide the reader with a general description of various commonly used in vivo rodent AP models, to discuss their strengths and weaknesses, and to discuss how the various models relate to human disease. In addition to this summary, we will make a number of “opinion-based” comments regarding the use of these models and the interpretation of the results obtained. We will suggest (a) which models are, in our opinion, most appropriate for addressing specific AP-related questions; (b) why we believe investigators should consider using more than one type of model for their studies; (c) how the severity of observed changes might best be quantitated; and (d) how discordant results obtained from studies employing more than one type of model might best be interpreted. Our review will focus entirely on issues related to non-alcohol related AP and we will not discuss models of alcohol-induced AP or models of chronic pancreatitis. In addition, we will not discuss issues related to hereditary pancreatitis, models of genetically-induced AP, and particularly those models that have only recently been described and have not been extensively used or validated. Our review will focus on in vivo models but, since most of the commonly used in vivo models can be replicated for ex vivo use, we will also briefly discuss some of these ex vivo systems.
Types of Models
The ideal experimental AP model would be technically simple to create, minimally invasive, reproducible, well characterized, inexpensive and resemble the human disease with respect to its triggering event, pathologic morphology, pathophysiology, disease course and response to treatment (20). Needless to say, none of the existing AP models fulfills all of these criteria. Therefore, it is not surprising that none of the pancreatitis models is universally used and that the pathophysiology of AP still remains poorly understood.
Noninvasive Models of Acute Pancreatitis
In general, noninvasive AP models are relatively simple and inexpensive to create and, therefore, their use has become quite popular. However, none of them are relevant to the human disease with respect to their etiology (i.e. their triggering event). Here, the most commonly used noninvasive models induced by (a) supramaximal stimulation with secretagogues, (b) feeding a CDE diet, and (c) administration of basic amino acids will be discussed in detail.
Secretagogue-induced models
Cerulein is a more stable analog of the gastrointestinal secretagogue hormone cholecystokinin (CCK). In maximally stimulating concentrations, cerulein or CCK causes the release of digestive enzymes from pancreatic acinar cells (59). However, as initially shown by Lampel and Kern (31), in higher, supramaximally stimulating concentrations, CCK or cerulein inhibit digestive enzyme secretion, cause premature intrapancreatic proteolytic enzyme activation, and induce AP. In rats, cerulein causes mild edematous AP (31), but in mice, it causes more severe, necrotizing AP (43). In both species, the disease is transient and self-limited. Therefore, it is not surprising that mortality in cerulein-induced pancreatitis is non-existent in rats and negligible in mice. This may be due to a mild pulmonary injury in cerulein-induced AP which has been shown to resemble the early stages of adult respiratory distress syndrome in human AP (73).
Cerulein can be administered parenterally via intraperitoneal (i.p.), i.v. or subcutaneous injections (37). The i.v. route, which allows for continuous cerulein administration (at doses of 5-50 mg/kg/h), is thought to be the best way of administering the hormone to rats; however, it is not commonly used due to the requirement of central venous cannulation and anesthesia. For this reason, the cerulein-induced model is usually elicited by the i.p. or subcutaneous administration of several (4-12) hourly doses of the secretagogue. In rats and mice, pancreatic injury (as manifest by trypsinogen activation, nuclear factor-κB activation, and vacuole formation) evolves within an hour of the start of cerulein administration and the peak of histological changes (interstitial edema, inflammation, and acinar cell injury/death) is noted 3-6 hours after the start of secretagogue administration. By 24 hours after the start of supramaximal secretagogue stimulation, these changes begin to resolve, and one week later, the pancreas appears to be morphologically normal. The severity of the disease can be adjusted by varying the dose and number of cerulein injections. Similar to the clinical characteristics of human AP, the severity of cerulein-induced AP is more pronounced in aged mice, which exhibit higher mortality rates (47). An advantage of using cerulein administration to animals to elicit AP is the fact that isolated acini can also be exposed to supramaximally stimulating concentrations of cerulein and, in this way, in vivo studies can be complemented by ex vivo studies under more completely controllable conditions.
A similar secretagogue-induced model has been developed to induce moderate AP in rats employing a single i.p. injection of 25 to 250 µg/kg carbachol, a cholinergic agonist (9, 23). This model leads to edematous pancreatitis characterized by hyperamylasemia and cellular injury. The animals also develop diarrhea and excessive lacrimation and salivation. At the higher doses (250 µg/kg) the rats die, presumably due to pulmonary edema. The pancreatitis induced by cholinergic agonists may be considered a model for the pancreatitis that results from scorpion venom (3, 65).
The secretagogue-induced models are the most commonly used and best characterized AP models in rodents, despite the questionable clinical relevance of their initiating event. In rodents, cerulein acts through the CCK receptors (in their low affinity state) on acinar cells. However, it is debatable whether human acinar cells express any CCK receptors. Murphy et al. (41) found that isolated human acinar cells respond to CCK by manifesting cytosolic Ca2+ signaling, activating mitochondrial function, and stimulating digestive enzyme secretion. In contrast, Ji et al. (26) and Miyasaka et al. (39) were unable to obtain any functional responses. It is likely that even if human acinar cells express CCK receptors, their expression level is markedly lower compared to that of rodents.
Choline-deficient ethionine-supplemented (CDE) diet-induced model
This model was originally described by Lombardi et al. (35). It is the least invasive of all the AP models since it requires no injections and no anesthesia or surgery. In this model, young female mice are fed a choline-deficient diet that is supplemeted with 0.5% ethionine (CDE diet) and they develop acute hemorrhagic pancreatitis with fat necrosis that occurs throughout the peritoneal cavity. They also develop a poorly characterized but very prominent liver injury. Feeding only an ethionine-supplemented diet can lead to edematous pancreatitis (17). The onset of CDE diet-induced AP is variable, but it usually takes approximately 2 to 3 days of diet administration for the disease to develop. Systemic effects such as acidosis, hypoxia and hypovolemia can also be observed. If the diet is fed ad libitum, it is usually lethal after 4 or 5 days of administration. Consumption of the CDE diet (and, thus, the severity of diet-induced pancreatitis) can vary considerably between groups of animals. Some mice would rather die than eat the CDE diet and this possible variability in CDE diet consumption can severely complicate the design of experiments. Careful record keeping and the use of large numbers of animals in each experimental group are important. Control and experimental groups of animals should be composed only of young, age-matched female mice given aliquots of the same CDE diet preparation and diet consumption should be controlled so that each animal consumes 3 g of diet/day. Because mice are frequently cannibalistic, animal mortality should be determined by counting the number of living mice at each time rather than counting the number of dead mice in each cage. The mortality of the model can be varied by changing the duration of CDE diet administration (44). Gilliland and Steer (21) were able to reduce the severity of AP by modifying the feeding protocol. Homogeneity and reproducibility of CDE diet induced AP depend on controlling the sex, age, weight and CDE diet-intake of the mice. According to the most commonly used protocol, CD-1 female mice (11-13 g) are starved for 1 day (to promote subsequent CDE diet consumption), then fed 3 g/mouse of the CDE diet on each of the subsequent 3 days, and then fasted for another day before being placed back on a regular laboratory diet. Young mice are affected more severely than adult mice, and females more than males (36). Estradiol-treatment of male mice sensitizes the animals to CDE diet-induced AP, so estrogens are likely to play an important role in the sex-specific nature of this model (55). Systemic signs, such as ascites, hypovolemia, acidosis and hypoxia accompany the local pancreatic inflammation (44). Unfortunately, the diet also affects the liver as well as the central nervous system and these non-pancreatic effects contribute to multiple organ failure and, eventually, to the death of the animals. Therefore, this model is not ideal for studying multiple organ distress syndromes because it can trigger those syndromes by mechanisms which are unrelated to the severity of AP. Another drawback of this model is that it elicits a severe disturbance of glucose metabolism (i.e. hypoglycemia) (71). Also, despite the severe acinar necrosis, the incidence of pancreatic infection is low (i.e. 3 % in survivor and 8% in non-survivor mice) (57, 70).
Acute pancreatitis induced by administration of basic amino acids
Whereas amino acids are essential components of the body, large i.p. doses of L-arginine (24, 40, 69), L-ornithine (54) and L-lysine (8), but not L-histidine (7) have been shown to induce severe acute necrotizing pancreatitis. Most commonly L -arginine in doses of 2.5-5.0 g/kg are used to induce experimental AP in rats. Two i.p. doses of 2.5 g/kg given at an interval of one hour do not produce as severe AP as a single 5 g/kg dose. For a long time, it was thought that this model of experimental pancreatitis could only be elicited in rats, but recently Dawra et al. (14, 16) have shown that using even higher L-arginine doses (2x4 g/kg) can also induce AP in mice. In the case of L-lysine, the disease evolves with the formation of vesicular structures, recently identified as damaged mitochondria (8), within pancreatic acinar cells. Pancreatic edema and necrosis followed by inflammation has been observed in this model and peripancreatic necrosis as well as ascites can also be observed, but hemorrhage is not a typical feature of this model.
Basic amino acids induce selective acinar cell damage without any apparent effect on duct and islet cells. Thus, the basic amino acid-induced models seem to morphologically resemble human necrotizing pancreatitis in which, as noted by Kovalska et al. (30), nerves, major ducts and islets are not markedly affected. One important drawback of the basic amino acid-induced models is that extrapancreatic complications (such as pulmonary insufficiency) due to AP are mild in these models (e.g. see 20). For this reason, these models are not suitable for studies focused on the pathophysiology of extrapancreatic AP-associated events. This may also explain the low mortality in this model. In mice, the effective and toxic/lethal doses of basic amino acids are very close to each other and toxicity, which is frequently lethal, is most likely to be due to metabolic effects of the basic amino acids themselves rather than to the associated AP. Rats and mice become lethargic soon after the i.p. injection of amino acids and it takes several hours for the animals to recover from this phase. The mortality associated with the L-arginine model in mice has been reported to be 5-7% (16). I.p. administration of more than 5 g/kg L-arginine, 3 g/kg L-ornithine or 2 g/kg L-lysine causes high mortality that occurs very shortly after the basic amino acid injection in rats, independent of AP. Notably, sensitivity of rats to basic amino acids seems to be strain- and age-specific and - with the exception of L-arginine - it is difficult to obtain a graded response. Bohus et al. (10) found a subset of Sprague-Dawley rats that responded weakly to the injection of 4 g/kg L-arginine. Similarly, we found that this also occurred with the i.p. administration of 2 g/kg L-lysine (8).
As with many AP models, the clinical relevance of basic amino acid-induced pancreatic injury remains questionable. Saka et al. (58) reported a 16-year-old male patient who was suspected to have arginine-induced AP after taking 500 mg arginine a day for 5 months. However, this is not a very high dose and the route of intake was oral, not i.p. Rats that (accidentally) receive the L-arginine injection into their bowels do not develop AP. Therefore, we believe that arginine intake was unlikely the cause of AP in this patient.
Invasive Models of Acute Pancreatitis
The most commonly used invasive AP models build upon the ”common channel theory” first proposed by Opie in 1901 (48). According to Opie’s theory, when a gallstone is impacted in the papilla of Vater (at the end of the common biliopancreatic channel), it can create a common channel up-stream to the impacted stone and, as a result, bile can retrogradely flow into the pancreatic duct and initiate AP. However, there are numerous arguments against this theory (67). Among these objections is the fact that, in many individuals, the common channel is so short that a gallstone (or the edema around a stone) impacted in the papilla of Vater would also obstruct the pancreatic duct and that phenomenon would prevent the outflow of pancreatic juice from the pancreatic duct. However, it would also prevent bile reflux into the pancreatic duct. Another objection frequently raised regarding the common channel theory is that pancreatic duct pressure normally exceeds bile duct pressure, so in cases of stone-induced distal obstruction, pancreatic juice would be expected to reflux into the bile duct while bile reflux into the pancreatic duct would likely be prevented. However, perhaps the most compelling of the objections to the common channel theory is the fact that perfusion of the pancreatic duct with bile, under normal pressures, does not cause pancreatic damage unless ductal pressure is also increased. In spite of these various objections, the so-called “common channel theory” continues to be an attractive explanation for the frequently noted association between biliary tract stone passage and the onset of acute biliary pancreatitis.
Besides their attractive feature of possibly mimicking the triggering event of human biliary pancreatitis, the invasive AP models require anesthesia of the animal which, by itself can also be challenging. Also postoperative problems, such as infections and difficulties in maintaining nutrition, can make the interpretation of results difficult.
Retrograde ductal infusion
The retrograde infusion of substances (e.g. bile acids, enterokinase, trypsin) intro the pancreatic duct of an animal via the ampulla of Vater is known to induce pancreatic inflammation. In fact, AP is also observed in about 5% of cases after the retrograde infusion of an X-ray contrast material used for endoscopic retrograde cholangiopancreatography in humans (64).
One of the most commonly used retrograde ductal infusion protocols was that described by Aho et al. (1) in rats. Their model uses 3 to 5% sodium taurocholate (1 ml/kg) which is infused at a rate of 0.1 ml/min. Sodium bile salts used by others include glycodeoxycholate, taurodeoxycholate, chenodeoxycholate and taurolithocholic acid 3-sulfate. Recently, the retrograde ductal infusion technique has also been adapted for mice (32, 52, 72, 74, 76). In either rats or mice, the severity of the disease can be controlled by altering the concentration, volume and infusion pressure of the injected bile acid. Administration of 3% sodium taurocholate causes mild pancreatitis with no mortality over 72 hours, 5% sodium taurocholate causes a more severe disease and a higher mortality. The disease induced by retrograde ductal bile acid infusion is also associated with extrapancreatic organ involvement but, unfortunately, creation of the model requires both anesthesia and a surgical procedure and that may make interpretation of its extrapancreatic effects very difficult. This is particularly true when studies are designed to evaluate acute lung injury after induction of pancreatitis by retrograde bile acid infusion into the pancreatic duct. Unlike the noninvasive necrotizing AP models, secondary pancreatic infections are more common following retrograde duct infusion. However, this higher infection rate may, at least partly, be the result of exogenously introduced organisms.
Technically, it is difficult to control for constant pressure of infusion and hence to produce a standard degree of pancreatic injury. Use of pumps makes the procedure more controllable/standardized. Uncontrolled pressure-related pancreatic damage should be avoided as it causes variation in the severity of AP. The retrograde ductal injection of substances such as bile acids will result in AP with a focal distribution mainly affecting the pancreatic head, but not the tail (32). This must always be kept in mind when sampling the pancreatic tissue for analysis. On the other hand, the disease that does develop in this model tends to have a patchy, non-homogeneous distribution throughout the affected portion of the gland which is similar to that seen in humans with AP.
In general, retrograde ductal infusion-induced pancreatitis is elicited in anesthetized rodents via a small laparotomy and transduodenal, transpapillary, cannulation of the pancreaticobiliary ductal system. After low pressure infusion of a solution containing selected bile acids or other suspected pancreaticotoxic agents, the cannula is removed and the animal allowed to recover. Control animals undergo infusion with only saline and this elicits only mild and transient pancreatic edema which resolves, entirely, within 24 hours. Pancreatitis, when it develops following bile acid infusion, usually evolves slowly and reaches its peak severity over the initial 12-24 hours. It is characterized by patchy pancreatic injury/necrosis, pancreatic inflammation, pancreatic edema, and intrapancreatic activation of digestive enzyme zymogens. Left untreated, bile acid infusion-induced pancreatitis in rodents resolves spontaneously over the subsequent week and, thereafter, the pancreas appears morphologically normal.
Schmidt et al. (61) modified the rat duct infusion model by combining the short-term pressure and volume-controlled retrograde injection of low concentrations (5-10 mM) of sodium glycodeoxycholic acid with the i.v. infusion of cerulein (5 μg/kg/h for 6 h). This so-called “Boston Model” is thought to resemble human necrotizing AP in many ways including the fact that it triggers both local and systemic changes (20). However, the triggering event does not resemble human AP.
Recent advances in the research of biliary AP models was reviewed by Wan et al. (72). One potentially important study in this field demonstrated that biliary AP may be a receptor-mediated disease (51). The G protein-coupled bile acid receptor-1 (Gpbar1) is expressed in the apical membrane of acinar cells. Its genetic deletion significantly reduces biliary, but not secretagogue-induced experimental AP (51).
Closed duodenal loop
The closed duodenal loop model was originally described by Pfeffer et al. (53) in dogs. In this model, the duodenum is obstructed by the placement of two ligatures, one just beyond the pylorus and the second placed just beyond the point of bilio-pancreatic inflow. This creates a closed intestinal segment that communicates with the biliopancreatic duct. Bile is excluded by ligating the biliary duct and gastric outflow is re-established by constructing a gastrojejunostomy. Closing the duodenal lumen both proximal and distal to the papilla of Vater will result in the reflux of duodenal contents into the biliopancreatic duct and this will cause AP. Acute pancreatitis induced using this approach is quite variable and difficult to control. Most investigators note that given the pancreatic necrosis, inflammation is mild.
The closed duodenal loop technique has also been adapted for rats by Nevalainen and Seppä (42). In their model, an intraduodenal tube was placed into the lumen of the intestine, prior to its ligation, to maintain duodenal continuity. Within 24 hours, a variable degree of hemorrhagic AP is observed. Increased serum amylase activity, pancreatic edema, acinar cell necrosis, and haemorrhage as well as intra-abdominal fat necrosis, and the accumulation of ascitic fluid containing high activity of amylase is detected. Chetty et al. (12) modified the latter model by instilling infected bile into the closed duodenal loop under pressure and this resulted in a more reproducible AP. Orda et al. (49) injected a combination of sodium taurocholate and trypsin into the permanently occluded duodenal loop, and this resulted in a mortality of 45% within one week. Dickson et al. (18) analyzed three closed duodenal loop model variants both histologically and bacteriologically. Histological studies showed that the resulting AP is usually mild to moderate and it is severe only in association with sepsis. Bacteriological studies revealed gross infection as a major complication. Interestingly, in the closed duodenal loop model, pancreatic necrosis is not necessary for infection to occur (70). This and other observations made with the closed duodenal loop model have suggested, to many, that the major cause of injury in this model may be ischemic necrosis of the duodenum rather that primary AP and the closed duodenal loop model is rarely used by investigators currently.
Duct obstruction/ligation
Interestingly, simple ligation of the pancreatic duct does not usually induce severe AP in most animals. The major exception to this generalization seems to be the American opossum (62). Short-term obstruction of the rat pancreatic duct results in mild interstitial pancreatic edema and hyperamylasemia (46) while longer exposure to ligation results in atrophy of the rat exocrine pancreas with very mild or no inflammation (13). However, combining duct obstruction with stimulation of pancreatic secretion can induce AP in rats.
The biliopancreatic ductal system of the opossum resembles that of humans – i.e. the biliary and pancreatic ducts merge several cm before the combined duct opens into the duodenum. Duct ligation in the opossum causes severe necrotizing hemorrhagic AP which evolves over a period of several days. This pancreatitis is also associated with lung injury (34, 62). All of the animals die within 14 days after ligation. Experiments performed in American opossums suggest that bile may not be necessary to induce AP, since the obstruction of the separate pancreatic duct produces pancreatitis which is similar in severity compared to that which is seen after simple ligation of the combined biliopancreatic duct (34). Unfortunately, the opossum model has several limitations which are mainly related to the animals being studied (66). The opossums used for these experiments are wild and they are collected from the wilderness. They are difficult to maintain and handle; of course this also means that the animals are not inbred and, as a result, there are large inter-animal variations of results. The animals also need to be preconditioned prior to their use for experiments since they are under considerable stress while in the early days of captivity. Opossums are often infected with parasites and thus require anti-helminthic treatment. Furthermore, since they frequently acquire acute bacterial endocarditis in captivity, they should be given prophylactic antibiotics before use. Because of the difficulties involved in obtaining, handling, and caring for opossums as well as the considerable animal-to-animal variations in results, the opossum model of duct ligation-induced pancreatitis is, currently, only infrequently used.
Choosing the “Best” Model of Acute Pancreatitis
Basic Requirements: Cost and Severity
Clinical material including pancreatic tissue is rarely available for study during the early phases of AP. Therefore, most investigations focused on mechanistic issues related to AP must be performed using animal as well as ex-vivo models of the disease. The two basic requirements of a useful animal model of AP are both pragmatically based – most of the commonly used pancreatitis models are (a) elicited in rodents (rats and mice) which are cheap, easy to handle, readily available, and subject to genetic manipulation; and (b) characterized by moderate to severe degrees of pancreatic injury. As noted earlier in this review, the severe form of clinical AP is responsible for almost all of the clinical pancreatitis-related morbidity and mortality while mild clinical pancreatitis is, by and large, a transient and self-limited disease with little or no morbidity or mortality.
The Good and the Bad about Rodent Models of Acute Pancreatitis
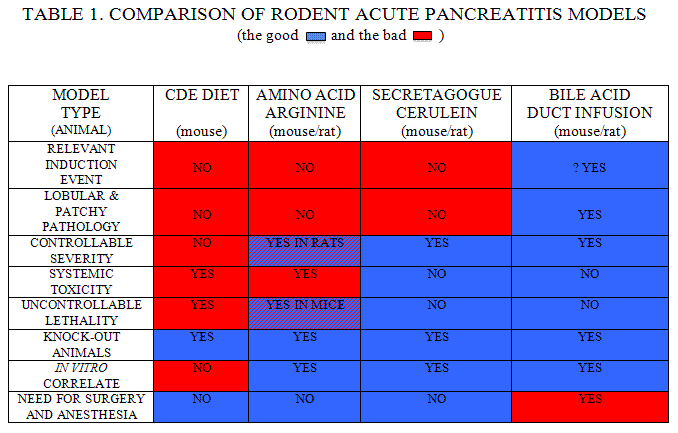
The four most frequently utilized types of rodent AP models are (a) the CDE diet-induced model; (b) the basic amino acid-induced models; (c) the secretagogue (cerulein)-induced models; and (d) the retrograde duct infusion models. We will discuss the relative merits of these four types of models and identify those that are best suited for use in studies probing basic issues related to AP. In this discussion, we will evaluate these models in terms of 8 separate criteria which, in our opinion, would each have an important impact on the overall value and utility of the models. Those criteria (Table 1) are as follows:
- Is the pancreatitis-triggering event in the model similar to the event(s) believed to trigger clinical AP (i.e. passage of biliary tract stones, abuse of ethanol, exposure to certain drugs, expression of certain mutated genes, performance of endoscopic retrograde cholangiopancreatography, etc.)?
- Do the pathologic changes elicited in the model replicate those that are noted in clinical pancreatitis (i.e. is the distribution of pancreatic injury lobular and patchy as it is in clinical AP or is it diffuse and homogeneous throughout the gland)?
- Is the severity of pancreatic injury/inflammation controllable by the investigator and is the magnitude of pancreatic injury significant so that increases or decreases in response to experimental interventions can be identified?
- Is the model associated with significant systemic toxicity caused by the eliciting event(s) which could confound the interpretation of specific intrapancreatic and systemic changes associated with the model?
- Is the magnitude of lethality in the model controllable by the investigator so that changes resulting from specific interventions can be detected and statistically evaluated?
- Can the model be elicited in mice so that the many mouse knock-out or otherwise genetically modified strains can be used in mechanistic studies?
- Is there an ex vivo correlate of the model so that in vitro studies can be performed under conditions which are more controllable than those present when only in vivo studies are possible?
- Does the model require surgery (which may be difficult) or anesthesia (which may, itself, trigger a variety of non-specific systemic changes thus complicating data interpretation and limiting the types of studies which are possible).
Strengths and Weaknesses of Individual Models
The results of our “model comparison” in terms of these criteria can be summarized as follows (see Table 1):
CDE diet model in mice
The major strengths of this model are its ability to be induced in mice, thus enabling the use of genetically modified mouse strains, and the fact that it is a non-invasive model which requires neither anesthesia nor a surgical procedure. Unfortunately, these attractive features are more than offset by a number of weaknesses. These include the fact that (a) ingestion of the CDE diet is not clinically associated with the triggering of clinical AP; (b) CDE diet-induced pancreatic injury in mice is diffuse rather than lobular or patchy as is the case in clinical pancreatitis; and (c) there is no ex vivo correlate for this in vivo model. An even more critical flaw in this model is the fact that CDE diet-induced pancreatitis in mice has a severity which is highly dependent upon the age, sex, and size of the mouse and closely related to the amount of the CDE diet ingested as well as the duration of diet administration. To adjust for these animal-to-animal causes of varied pancreatitis severity, the investigator is forced to use large numbers of age and sex-matched mice, each simultaneously exposed to a standard amount of the same CDE diet preparation. This can be extremely cumbersome even when only wild type animals are being used and can represent an insurmountable obstacle when genetically modified mice are being used. Another critical flaw of the CDE diet induced model results from the fact that the diet is associated with the induction of severe and uncontrollable non-pancreatic injury (primarily central nervous system injury and severe liver toxicity) which probably account for most of the diet-induced mortality of this model. These non-pancreatitic injuries also preclude the use of this model for studies focused on quantitation of pancreatitis-associated systemic phenomena such as pancreatitis-associated lung and/or renal injury.
Basic amino acid (L-arginine)-induced model in mice and rats
Like the CDE diet-induced model, the models of AP induced by administration of basic amino acids such as arginine are attractive because they are non-invasive and, therefore, do not require anesthesia or performance of a surgical procedure. At least in the case of L-arginine, this form of pancreatitis can be elicited in mice, thus enabling the use of genetically modified animals. However, in mice there is a very narrow margin of error when arginine is used – i.e. the dose required to elicit pancreatic injury is only slightly less than the toxic dose for mice and, as a result, control arginine-treated groups of animals are needed to establish the fact that otherwise untreated animals do, in fact, manifest non-lethal pancreatic injury. Other possible flaws in the L-arginine-induced models include the generalized, as opposed to patchy, distribution of pancreatic injury and the systemic toxicity of pancreatitis-eliciting doses of the basic amino acid which may preclude the use of this model in studies designed to explore issues related to systemic events (e.g. lung injury) in AP. Clearly, although exposure of acini to toxic concentrations of L-arginine results in cell damage (25), this is not a likely triggering event in clinical pancreatitis. However, recent in vitro studies by our group (8) indicate that basic amino acids such as L-lysine can elicit mitochondrial injury in acinar cells which is known to be an early event in secretagogue-induced and bile acid-induced pancreatitis. Thus, in this regard, the basic amino acid-induced model may, in fact, be triggered by a clinically relevant mechanism (i.e. mitochondrial injury) but further studies will be needed to establish this.
Cerulein models in rats and mice
These models are elicited by administration of supramaximally stimulating doses of the pancreatic secretagogue CCK or its analog cerulein. They are attractive models for a number of reasons including the fact that they are associated with easily controllable and reproducible AP severity with no systemic toxicity or uncontrollable lethality. In addition, they are non-invasive in the sense that they require no anesthesia or surgical procedure and, as a result, they are ideally suited for studies exploring systemic events that occur during or after the onset of pancreatitis. They can be used with genetically engineered mice and ex vivo studies exploring the effects of supramaximal secretagogue stimulation on isolated acinar cells are easily performed. The major flaws of these models, however, are (a) the fact that they are elicited by an event (supramaximal secretagogue-stimulation) which is unlikely to contribute to the onset of clinical pancreatitis and (b) the observation that CCK or cerulein-induced pancreatitis is characterized by diffuse, homogeneous evidence of pancreatic injury rather than the patchy pancreatic injury which typifies clinical pancreatitis.
Retrograde bile acid infusion into the pancreatic duct of rats or mice
These models have, in the past, been extensively used with rats or larger animals but, as recently shown by our group (32), they can also be modified for use with mice. The most attractive features of these models are (a) the fact that their induction mimics an event (i.e. bile acid reflux into the pancreatic duct) which has been proposed to be the mechanism which triggers the most common form of clinical, non-alcohol related AP (i.e. biliary pancreatitis) and (b) the fact that the pancreatic injury which occurs in these models has a patchy or lobular distribution which closely resembles the distribution of pancreatic injury that is noted in clinical pancreatitis. Other attractive features of these models include their easily controllable and reproducible severity (especially in mice) and the fact that they are not associated with systemic toxicity or uncontrollable lethality. In addition, ex vivo studies in which isolated acini are exposed to bile acids can easily be performed and, thus, the potential mechanisms by which bile acids trigger pancreatic injury and pancreatitis in vivo can be explored under more controllable ex vivo conditions. The major weakness of these models, however, stems from the fact that they require anesthesia and a laparotomy for their induction. In mice, that may be difficult but, in both mice and rats, it limits the utility of these models because the pulmonary and other systemic effects of anesthesia and a laparotomy make it difficult, if not impossible, to isolate the effects of AP from the effects of the surgery on these changes.
Quantitating the Severity of Rodent Experimental Acute Pancreatitis
It is often critical that the experimentalist be able to reliably quantitate both the severity of the model being employed and the response of that severity to experimental manipulation. Each of the models is characterized by hyperamylasemia/hyperlipasemia, pancreatic edema, intrapancreatic activation of digestive enzyme zymogens, pancreatic inflammation, and morphological changes suggestive of acinar cell injury/death. Hyperenzymemia can be easily quantitated by measuring the activity of amylase and/or lipase in circulating blood. While it is standard practice to demonstrate that the model being used results in elevated serum amylase and/or lipase activity, the magnitude of that hyperenzymemia is not believed to be an indicator of pancreatitis severity.
The other characteristics of AP mentioned above are believed to be individual and separable indicators of pancreatitis severity although, to some degree, they may be inter-observer dependent.
In our opinion, they should be separately measured and reported. Grading them on a 1-4+ scale and combining those scores to calculate a so-called “pancreatitis severity score” may, we believe, be misleading because it presumes that the individual parameters of AP severity are interchangeable (e.g., that 1 unit of pancreatic inflammation is equal to 1 unit of pancreatic edema or 1 unit of acinar cell injury/death). Pancreatic edema can be objectively quantitated by measuring pancreatic water content [i.e. (wet weight minus dry weight) divided by wet weight] (22) although some investigators chose to quantitate edema morphologically, grading it as being 1-4+.
We favor the former method because, in our opinion, it is more objective. Intrapancreatic zymogen activation can be fluorimetrically quantitated by measuring trypsin and/or chymotrypsin activity in pancreas homogenates using enzyme-specific fluorescent substrates (22, 26) while pancreatic inflammation can be quantitated by measuring the myeloperoxidase activity in pancreas homogenates (15). Some investigators have preferred to monitor inflammation morphologically by quantitating inflammatory cells within pancreas tissue samples. Because of its ease and, in our opinion, greater reliability, we prefer the former method for quantitation of pancreatic inflammation. Acinar cell injury/death is conventionally monitored morphometrically in tissue samples of unknown identity (i.e. in a “blinded” fashion), by quantitating the fraction of acinar cells which appear to be injured or dead (27) but it is important to recognize that morphological distinction between injured and dead acinar cells is usually not possible.
The Best Model to Use May Depend Upon the Questions Being Asked
None of the rodent models of AP is perfect and each has its own strengths/weaknesses. In our opinion, the best models for studies probing the very early pancreatic cell biological mechanisms responsible for the triggering of AP are those models that are elicited by retrograde ductal infusion of bile acids in mice or rats. That judgment is based on the belief that those models are (a) triggered by a mechanism which may also be relevant to the triggering of clinical pancreatitis and (b) characterized by a distribution of pancreatic changes that resembles the distribution observed in clinical pancreatitis (i.e. patchy rather than homogeneous and diffuse). On the other hand, for studies focused on the more down-stream pancreatic events that might be expected to be similar regardless of the initial, triggering events, we believe that it would be appropriate to also employ the cerulein-induced models and/or the basic amino acid-induced models. We also believe that, because of their ease of use, non-invasive induction, and lack of systemic toxicity, the cerulein-induced models are those which are most appropriate for studies focused on non-pancreas related events associated with pancreatitis such as acute lung injury or the systemic inflammatory response syndrome (63, 75). While inducing either mild (rat) or severe (mouse) pancreatitis, these cerulein-induced models avoid the need for anesthesia and a surgical procedure which could confound the results by causing pulmonary and systemic changes even in the absence of AP. In our opinion there is little or no on-going justification for the use, in pancreatitis-related studies, of (a) the CDE diet induced model; (b) the pancreatic duct obstruction models; or (c) the closed duodenal loop model.
The Value of Performing Studies Using More Than One Rodent Model of Acute Pancreatitis and Interpretation of Discordant Results
Many, but certainly not all, of the AP-related studies reported to date have been performed using 2 or more experimental models of and, for the most part, the results obtained when multiple models have been tested have been consistent – i.e. identical interventions in different models have resulted in similar changes in pancreatitis severity. This has generally been interpreted as indicating that the phenomena being studied are relevant to the general issue of AP rather than being idiosyncratic manifestations of model-specific phenomena. Recently, however, a series of studies using 2 different mouse models of AP (i.e. the mouse cerulein-induced model and the mouse duct infusion-induced model) were reported that evaluated the effects, on pancreatitis severity, of 2 interventions -- genetic deletion and pharmacological inhibition of Protease-Activated Receptor-2 (PAR2). The studies showed that genetic deletion or pharmacological inhibition of PAR2 had dramatically different effects on the severity of the 2 models – i.e. they worsened the severity of cerulein-induced pancreatitis but they lessened the severity of duct infusion-induced pancreatitis (33, 38). These unexpected and surprising results indicating that PAR2 exerts model-specific effects on the severity of AP were interpreted to indicate that the severity of the two models are differentially regulated by one or more PAR2-sensitive mechanisms. In addition to demonstrating the value of performing pancreatitis studies using 2 or more dissimilar models of the disease, these studies also raised an additional important question, how should the investigator interpret the relevance of results to the clinical situation when studies using multiple models yield discordant results? When and if this should occur, we believe that the guide to clinical relevance should be the model which most closely resembles clinical pancreatitis. In this case, the duct infusion-induced model because (a) in contrast to cerulein-induced pancreatitis, it is triggered by a mechanism which may replicate the events that trigger clinical pancreatitis and (b) it is characterized by a distribution of pancreatic injury which is similar to the clinical pathology (i.e. variable and patchy) whereas, in cerulein-induced AP, pancreatic injury is diffuse and homogeneous.
Acknowledgements
Our research is supported by the National Development Agency (TÁMOP-4.2.2.A-11/1/KONV-2012-0035, TÁMOP-4.2.2-A-11/1/KONV-2012-0052 TÁMOP-4.2.2.A-11/1/KONV-2012-0073), the Hungarian Scientific Research Fund (NF105758 to Z.R., NF100677 to P.H.), the Hungarian Academy of Sciences (BO 00174/10/5 to Z.R.), and the National Institutes of Health (DK091327 to M.L.S.).
References
- Aho HJ, Koskensalo SM, Nevalainen TJ. Experimental pancreatitis in the rat. Sodium taurocholate-induced acute haemorrhagic pancreatitis. Scand J Gastroenterol 15: 411-416, 1980. PMID: 7433903.
- Banerjee AK, Galloway SW, Kingsnorth AN. Experimental models of acute pancreatitis. Br J Surg 81: 1096-1103, 1994. PMID: 7953329.
- Bartholomew C. Acute scorpion pancreatitis in Trinidad. Br Med J 14:666-668, 1970. PMID: 5443968.
- Beger HG, Bittner R, Block S, Büchler M. Bacterial contamination of pancreatic necrosis. A prospective clinical study. Gastroenterology 91:433-438, 1986. PMID: 3522342.
- Beger HG, Rau B, Mayer J, Pralle U. Natural course of acute pancreatitis. World J Surg 21: 130-135, 1997. PMID: 8995067.
- Bernard C. Leçons de Physiologie Expérimentale Appliquée a la Médecine Editor Bailliere, J.-B. Paris, Vol. 2, 230-252, 1856.
- Biczó G, Hegyi P, Dósa S, Balla Z, Venglovecz V, Iványi B, Wittmann T, Takács T, Rakonczay Z Jr. Aliphatic, but not imidazole, basic amino acids cause severe acute necrotizing pancreatitis in rats. Pancreas 40: 486-487, 2011. PMID: 21412124.
- Biczó G, Hegyi P, Dósa S, Shalbuyeva N, Berczi S, Sinervirta R, Hracskó Z, Siska A, Kukor Z, Jármay K, Venglovecz V, Varga IS, Iványi B, Alhonen L, Wittmann T, Gukovskaya A, Takács T, Rakonczay Z Jr. The crucial role of early mitochondrial injury in L-lysine-induced acute pancreatitis. Antioxid Redox Signal 15:2669-2681, 2011. PMID: 21644850.
- Bilchik AJ, Zucker KA, Adrian TE, Modlin IM. Amelioration of cholinergic-induced pancreatitis with a selective cholecystokinin receptor antagonist. Arch Surg 125: 1546-1549, 1990. PMID: 2244806
- Bohus E, Coen M, Keun HC, Ebbels TM, Beckonert O, Lindon JC, Holmes E, Noszál B, Nicholson JK. Temporal metabonomic modeling of L-arginine-induced exocrine pancreatitis. J Proteome Res 7: 4435-4445, 2008. PMID: 18710274.
- Büchler M, Friess H, Uhl W, Beger HG. Clinical relevance of experimental acute pancreatitis. Eur Surg Res 24 Suppl 1:85-88, 1992. PMID: 1601028.
- Chetty U, Gilmour HM, Taylor TV. Experimental acute pancreatitis in the rat—a new model. Gut 21: 115-117, 1980. PMID: 6155312.
- Churg A, Richter WR. Early changes in the exocrine pancreas of the dog and rat after ligation of the pancreatic duct. A light and electron microscopic study. Am J Pathol 63: 521-546, 1971. PMID: 5581235.
- Dawra R, Sharif R, Phillips P, Dudeja V, Dhaulakhandi D, Saluja AK. Development of a new mouse model of acute pancreatitis induced by administration of L-arginine. Am J Physiol Gastrointest Liver Physiol 292: G1009-1018, 2007. PMID: 17170029.
- Dawra R, Ku YS, Sharif R, Dhaulakhandi D, Phillips P, Dudeja V, Saluja AK. An improved method for extracting myeloperoxidase and determining its activity in the pancreas and lungs during pancreatitis. Pancreas 37, 62-68, 2009, PMID:18580446
- Dawra R, Saluja AK. L-arginine-induced experimental acute pancreatitis. The Pancreapedia: Exocrine Pancreas Knowledge Base, 2012, DOI: 10.3998/panc.2012.6
- De Almeida AL, Grossman MI. Experimental production of pancreatitis with ethionine. Gastroenterology 20: 554-577, 1952. PMID: 14917176.
- Dickson AP, Foulis AK, Imrie CW. Histology and bacteriology of closed duodenal loop models of experimental acute pancreatitis in the rat. Digestion 34: 15-21, 1986. PMID: 3709998.
- Elder AS, Saccone GT, Bersten AD, Dixon DL. L-arginine-induced acute pancreatitis results in mild lung inflammation without altered respiratory mechanics. Exp Lung Res 37: 1-9, 2011. PMID: 21077777.
- Foitzik T, Hotz HG, Eibl G, Buhr HJ. Experimental models of acute pancreatitis: are they suitable for evaluating therapy? Int J Colorectal Dis 15: 127-135, 2000. PMID: 10954184.
- Gilliland L, Steer ML. Effects of ethionine on digestive enzyme synthesis and discharge by mouse pancreas. Am J Physiol 239: G418-26, 1980. PMID: 6159794.
- Grady T, Salja A, Kaiser A, Steer M. Edema and intrapancreatic trypsinogen activation preced glutathione depletion druing caerulein pancreatitis. Am J Physiol 271: G20-26, 1996, PMID 8760102.
- Grönroos JM, Laine J, Kaila T, Nevalainen TJ. Chronic alcohol intake and carbachol-induced acute pancreatitis in the rat. Exp Toxicol Pathol 46: 163-167, 1994. PMID: 7987075.
- Hegyi P, Rakonczay Z Jr, Sári R, Góg C, Lonovics J, Takács T, Czakó L. L‑arginine-induced experimental pancreatitis. World J Gastroenterol 10: 2003-2009, 2004. PMID: 15237423.
- Hu G, Shen J, Cheng L, Guo C, Xu X, Wang F, Huang L, Yang L, He M, Xiang D, Zhu S, Wu M, Yu Y, Han W, Wang X. Reg4 protects against acinar cell necrosis in experimental pancreatitis. Gut 60: 820-828, 2011. PMID: 21193457.
- Ji B, Bi Y, Simeone D, Mortensen RM, Logsdon CD. Human pancreatic acinar cells lack functional responses to cholecystokinin and gastrin. Gastroenterology 121: 1380-1390, 2001. PMID: 11729117.
- Kaiser AM, Saluja AK, Sengupta A, Saluja M, Steer ML. Relationship between severity, necrosis, and apoptosis in five models of experimental acute pancreatitis Am J Physiol 269:C1295-304, 1995. PMID: 7491921.
- Klöppel G. Acute pancreatitis. Semin Diagn Pathol 21: 221-226, 2004. PMID: 16273940.
- Klöppel G, Maillet B. Pathology of acute and chronic pancreatitis. Pancreas 8: 659-670, 1993. PMID: 8255882.
- Kovalska I, Dronov O, Zemskov S, Deneka E, Zemskova M. Patterns of pathomorphological changes in acute necrotizing pancreatitis. Int J Inflam 2012: 508915, 2012. PMID: 22611517.
- Lampel M, Kern HF. Acute interstitial pancreatitis in the rat induced by excessive doses of a pancreatic secretagogue. Virchows Arch A Pathol Anat Histol 373: 97-117, 1977. PMID: 139754.
- Laukkarinen JM, van Acker GJ, Weiss ER, Steer ML, Perides G. A mouse model of acute biliary pancreatitis induced by retrograde pancreatic duct infusion of Na-taurocholate. Gut 56: 1590-1598, 2007. PMID: 17591621.
- Laukkarinen JM, Weiss ER, van Acker GJ, Steer ML, Perides G. Proetase-activated receptor-2 exerts contrasting model-specific effects on acute experimental pancreatitits. J Biol Chem 283: 20703-20712, 2008. PMID: 18511423.
- Lerch MM, Saluja AK, Rünzi M, Dawra R, Saluja M, Steer ML. Pancreatic duct obstruction triggers acute necrotizing pancreatitis in the opossum. Gastroenterology 104: 853-861, 1993. PMID: 7680018.
- Lombardi B, Estes LW, Longnecker DS. Acute hemorrhagic pancreatitis (massive necrosis) with fat necrosis induced in mice by DL-ethionine fed with a choline-deficient diet. Am J Pathol 79: 465-80, 1975. PMID: 1094837.
- Lombardi B, Rao NK. Acute hemorrhagic pancreatic necrosis in mice. Influence of the age and sex of the animals and of dietary ethionine, choline, methionine, and adenine sulfate. Am J Pathol 81: 87-100, 1975. PMID: 1180334.
- Mayerle J, Sendler M, Lerch MM. Secretagogue (caerulein) induced pancreatitis in rodents. The Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2013.2
- Michael ES, Kuliopulos A, Covic L, Steer ML, Perides G. Pharmacologic inhibition of PAR2 with the pepducin P2pal-18S protects mice agains acute experimental biliary pancreatitis, Am J Physiol Gastrointest Liver Physiol 304:G516-526, 2013. PMID: 23275617
- Miyasaka K, Shinozaki H, Jimi A, Funakoshi A. Amylase secretion from dispersed human pancreatic acini: neither cholecystokinin a nor cholecystokinin B receptors mediate amylase secretion in vitro. Pancreas 25: 161-165, 2002. PMID: 12142739.
- Mizunuma T, Kawamura S, Kishino Y. Effects of injecting excess arginine on rat pancreas. J Nutr 114: 467-471, 1984. PMID: 6199486.
- Murphy JA, Criddle DN, Sherwood M, Chvanov M, Mukherjee R, McLaughlin E, Booth D, Gerasimenko JV, Raraty MG, Ghaneh P, Neoptolemos JP, Gerasimenko OV, Tepikin AV, Green GM, Reeve JR Jr, Petersen OH, Sutton R. Direct activation of cytosolic Ca2+ signaling and enzyme secretion by cholecystokinin in human pancreatic acinar cells. Gastroenterology 135: 632-641, 2008. PMID: 18555802.
- Nevalainen TJ, Seppä A. Acute pancreatitis caused by closed duodenal loop in the rat. Scand J Gastroenterol 10: 521-527, 1975. PMID: 1153948.
- Niederau C, Ferrell LD, Grendell JH. Caerulein-induced acute necrotizing pancreatitis in mice: protective effects of proglumide, benzotript, and secretin. Gastroenterology 88: 1192-1204, 1985. PMID: 2984080.
- Niederau C, Lüthen R, Niederau MC, Grendell JH, Ferrell LD. Acute experimental hemorrhagic-necrotizing pancreatitis induced by feeding a choline-deficient, ethionine-supplemented diet. Methodology and standards. Eur Surg Res 24 Suppl 1: 40-54, 1992. PMID: 1601023.
- Nordback I, Lauslahti K. Clinical pathology of acute necrotising pancreatitis. J Clin Pathol 39: 68-74, 1986. PMID: 3950033.
- Ohshio G, Saluja A, Steer ML. Effects of short-term pancreatic duct obstruction in rats. Gastroenterology 100: 196-202, 1991. PMID: 1700960.
- Okamura D, Starr ME, Lee EY, Stromberg AJ, Evers BM, Saito H. Age-dependent vulnerability to experimental acute pancreatitis is associated with increased systemic inflammation and thrombosis. Aging Cell 11: 760-769, 2012. PMID: 22672542.
- Opie EL. The etiology of acute hemorrhagic pancreatitis. Bull Johns Hopkins Hosp 12: 182-188, 1901.
- Orda R, Hadas N, Orda S, Wiznitzer T. Experimental acute pancreatitis. Inducement by taurocholate sodium-trypsin injection into a temporarily closed duodenal loop in the rat. Arch Surg 115: 327-329, 1980. PMID: 6153522.
- Pandol SJ, Saluja AK, Imrie CW, Banks PA. Acute pancreatitis: bench to the bedside. Gastroenterology 132: 1127-1151, 2007. PMID: 17383433.
- Perides G, Laukkarinen JM, Vassileva G, Steer ML. Biliary acute pancreatitis in mice is mediated by the G-protein-coupled cell surface bile acid receptor Gpbar1. Gastroenterology 138: 715-725, 2010. PMID: 19900448.
- Perides G, van Acker GJ, Laukkarinen JM, Steer ML. Experimental acute biliary pancreatitis induced by retrograde infusion of bile acids into the mouse pancreatic duct. Nat Protoc 5: 335-341, 2010. PMID: 20134432.
- Pfeffer RB, Stasior O, Hilton JW. The clinical picture of the sequential development of acute hemorrhagic pancreatitis in the dog. Surg Forum 8: 248-251, 1957. PMID: 13529599.
- Rakonczay Z Jr, Hegyi P, Dósa S, Iványi B, Jármay K, Biczó G, Hracskó Z, Varga IS, Karg E, Kaszaki J, Varró A, Lonovics J, Boros I, Gukovsky I, Gukovskaya AS, Pandol SJ, Takács T. A new severe acute necrotizing pancreatitis model induced by L-ornithine in rats. Crit Care Med 36: 2117-2127, 2008. PMID: 18594222.
- Rao KN, Eagon PK, Okamura K, Van Thiel DH, Gavaler JS, Kelly RH, Lombardi B. Acute hemorrhagic pancreatic necrosis in mice. Induction in male mice treated with estradiol. Am J Pathol 109: 8-14, 1982. PMID: 6181693.
- Rattner DW. Experimental models of acute pancreatitis and their relevance to human disease. Scand J Gastroenterol Suppl 219: 6-9, 1996. PMID: 8865463.
- Rattner DW, Compton CC, Gu ZY, Wilkinson R, Warshaw AL. Bacterial infection is not necessary for lethal necrotizing pancreatitis in mice. Int J Pancreatol 5: 99-105, 1989. PMID: 2664023.
- Saka M, Tüzün A, Ateş Y, Bağci S, Karaeren N, Dağalp K. Acute pancreatitis possibly due to arginine use: a case report. Turk J Gastroenterol 15: 56-58, 2004. PMID: 15264124.
- Saluja AK, Lerch MM, Phillips PA, Dudeja V. Why does pancreatic overstimulation cause pancreatitis? Annu Rev Physiol 69: 249-269, 2007. PMID: 17059357.
- Schmid SW, Uhl W, Kid M, Modlin IM, Buchler MW. Experimental models of acute pancreatitis and their clinical relevance. In: Buechler MW, Uhl W, Friess H, Malfertheiner P eds. Acute Pancreatitis. Novel Concepts in Biology and Therapy. Berlin: Blackwell Science, p. 51–62, 1999.
- Schmidt J, Rattner DW, Lewandrowski K, Compton CC, Mandavilli U, Knoefel WT, Warshaw AL. A better model of acute pancreatitis for evaluating therapy. Ann Surg 215: 44-56, 1992. PMID: 1731649.
- Senninger N, Moody FG, Coelho JC, Van Buren DH. The role of biliary obstruction in the pathogenesis of acute pancreatitis in the opossum. Surgery 99: 688-693, 1986. PMID: 2424109.
- Severgnini M Takahashi S, Rozo LM, Homer RJ, Kuhn C, Jhung JW, Perides G, Steer M, Hassoun PM, Fanburg BL, Cochran BH, Simon AR. Activation of the STAT pathway in acute lung injury. Am J Physiol Lung Cell Mol Physiol286: L1282-92, 2004. PMID:14729509
- Sherman S, Lehman GA. ERCP- and endoscopic sphincterotomy-induced pancreatitis. Pancreas 6: 350-367, 1991. PMID: 1713676.
- Sofer S, Shalev H, Weizman Z, Shahak E, Gueron M. Acute pancreatitis in children following envenomation by the yellow scorpion Leiurus quinquestriatus. Toxicon 29: 125-128, 1991. PMID: 2028471.
- Steer ML. Models for the study of pancreatitis. In: Souba WW, Wilmore DW, eds. Surgical Research. San Diego: Academic Press, p. 733-746, 2001.
- Steer ML, Perides G. Pathogenesis: How does acute pancreatitis develop? In: Domínguez-Muñoz JE ed. Clinical Pancreatology: For Practising Gastroenterologists and Surgeons. Blackwell Publishing Ltd, Oxford, UK, p. 10-26, 2007.
- Su KH, Cuthbertson C, Christophi C. Review of experimental animal models of acute pancreatitis. HPB (Oxford) 8: 264-286, 2006. PMID: 18333137.
- Tani S, Itoh H, Okabayashi Y, Nakamura T, Fujii M, Fujisawa T, Koide M, Otsuki M. New model of acute necrotizing pancreatitis induced by excessive doses of arginine in rats. Dig Dis Sci 35: 367-374, 1990. PMID: 2307082.
- van Minnen LP, Blom M, Timmerman HM, Visser MR, Gooszen HG, Akkermans LM. The use of animal models to study bacterial translocation during acute pancreatitis. J Gastrointest Surg 11: 682-689, 2007. PMID: 17468930.
- Virji MA, Rao KN. Acute hemorrhagic pancreatitis in mice: A study of glucoregulatory hormones and glucose metabolism. Am J Pathol 118: 162-167, 1985. PMID: 3881038.
- Wan MH, Huang W, Latawiec D, Jiang K, Booth DM, Elliott V, Mukherjee R, Xia Q. Review of experimental animal models of biliary acute pancreatitis and recent advances in basic research. HPB (Oxford) 14: 73-81, 2012. PMID: 22221567.
- Willemer S, Feddersen CO, Karges W, Adler G. Lung injury in acute experimental pancreatitis in rats. I. Morphological studies. Int J Pancreatol 8: 305-321, 1991. PMID: 1791317.
- Wittel UA, Wiech T, Chakraborty S, Boss B, Lauch R, Batra SK, Hopt UT. Taurocholate-induced pancreatitis: a model of severe necrotizing pancreatitis in mice. Pancreas 36: e9-21, 2008. PMID: 18376298.
- Zhang H, Neuhöfer P, Song L, Rabe B, Lesina M, Kurkowski MU, Treiber M, Wartmann T, Regnér S, Thorlacius H, Saur D, Weirich G, Yoshimura A, Halangk W, Mizgerd JP, Schmid RM, Rose-John S, Algül H. IL-6 trans-signaling promotes pancreatitis-associated lung injury and lethality. J Clin Invest 123:1019-1031, 2013 PMID: 23426178
- Ziegler KM, Wade TE, Wang S, Swartz-Basile DA, Pitt HA, Zyromski NJ. Validation of a novel, physiologic model of experimental acute pancreatitis in the mouse. Am J Transl Res 3: 159-165, 2011. PMID: 21416058.