
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2016.15
| Attachment | Size |
|---|---|
| 370.19 KB |
1. The role of the pancreatic acinar cell in acute pancreatitis:
Pancreatic necrosis, systemic inflammatory response syndrome, multiple organ failure and sepsis are characteristic of severe acute pancreatitis, which results in death of one in four patients and is without specific drug therapy (59, 62).As the pancreatic acinar cell is an initial site of injury (41, 59), commonly initiated by bile or ethanol excess, investigation of its behaviour in response to toxins that induce acute pancreatitis may determine critical mechanisms and importantly identify new drug targets. In view of the critical role of calcium signaling in normal stimulus-secretion and stimulus-metabolism coupling, and the long known toxicity of raised intracellular calcium, we proposed the hypothesis that prolonged elevations of cytosolic calcium is the key trigger of acute pancreatitis(83). Since that proposal over 20 years ago, increasing evidence has confirmed that sustained elevation of the cytosolic calcium concentration ([Ca2+]C) is a critical trigger for pancreatic acinar cell injury and necrosis that depends on store-operated calcium entry (SOCE) (12, 24, 36, 39, 63, 84).
2. The critical role of calcium entry in acinar cell injury:
Intracellular Ca2+ signals control normal secretion from pancreatic acinar cells but can become a critical trigger in pathogenesis. Physiological concentrations of acetylcholine (ACh) and cholecystokinin (CCK) generate repetitive elevations in [Ca2+]C within the cellular apical pole that elicit stimulus-metabolism coupling to generate adenosine triphosphate (ATP) from mitochondria and stimulus-secretion coupling to initiate exocytosis (61). Intermittently, global extension of short-lived signals throughout the cell are necessary for nuclear signalling contributing to transcription and translation (61). Elevations of [Ca2+]C are buffered in mitochondria, notably those surrounding the apical pole, with subsequent reuptake into the endoplasmic reticulum (ER) by sarcoER Ca2+ ATPase (SERCA) pumps and extrusion by plasma membrane calcium pump (PMCA) (Figure 1). In contrast, toxins such as bile acids (81), oxidative (70) and non-oxidative metabolites (12, 34) of ethanol and CCK hyperstimulation (10, 53) each elicit abnormal elevations of [Ca2+]C that are global and sustained. The abnormal elevations induce premature activation of intracellular enzymes, mitochondrial dysfunction, impaired autophagy, vacuolization and necrosis, all of which contribute to the pathogenesis of acute pancreatitis (11). Maintenance of these abnormal elevations depends on continued emptying of the ER Ca2+ store and activation of store-operated Ca2+ entry (SOCE) and Ca2+-release activated Ca2+ currents (CRAC) to replenish the ER store(24, 84). Ca2+chelation prevents zymogen activation and vacuolization through attenuation of Ca2+ overload in acinar cells in vitro (63, 67) and ameliorates the severity of acute pancreatitis in vivo (49).
Excessive Ca2+ release from intracellular stores occurs predominantly via inositol 1,4,5-trisphosphate receptor (IP3R) Ca2+ channels (26). The pancreatic acinar cell expresses all three subtypes of the IP3R in the apical region, close to the luminal membrane (21, 45, 46) but IP3R types 2 and 3 are predominantly responsible for physiological Ca2+ signaling and enzyme secretion (21). Stimuli such as CCK (7) the bile acid taurolithocholic acid 3-sulphate (TLCS) (23, 79), alcohol(25) and fatty acid ethyl esters (FAEEs)(12, 26) cause intracellular Ca2+ release in pancreatic acinar cells primarily via IP3Rs, an effect inhibited by double knockout of IP3R types 2 and 3 (21) or by caffeine (26, 53).
Since the discovery of the calcium entry channel ORAI1, ORAI1 has been shown to be the principal SOCE channel in the pancreatic acinar cell (45), opening of which is coordinated by stromal interaction molecules (STIM1 and STIM2), following decreases in ER calcium store concentrations (16, 24, 45, 50).

Figure 1. Ca2+ signaling in the pancreatic acinar cell depends on tight control of concentrations across the plasma membrane and within subcellular organelles. In resting conditions [Ca2+]c is ~10,000 fold lower than outside the cell, with Ca2+ stored mainly in the ER. Stimulus-secretion coupling operates by secretagogue-elicited GPCR activation to release second messengers that bind to inositol trisphosphate and ryanodine receptors on the ER, through which Ca2+ is released into the cytosol and mitochondria, initiating ATP production that provides energy for secretion. Ca2+ is cleared by reuptake into the ER and extrusion from the cell; the Ca2+ extruded is replenished via puncta forming between the ER and plasma membrane in response to low ER Ca2+ levels, allowing entry via ORAI channels. Pancreatitis toxins induce excessive ER Ca2+ release, initiating a vicious circle that overwhelms the cell (ER: endoplasmic reticulum; GA: Golgi apparatus; MT: mitochondria; NU: nucleus; ZG: zymogen granules).
GSK-7975A and CM_128 have been developed independently by GlaxoSmithKline (16, 24, 64) and CalciMedica (84) respectively to block ORAI1 channels, although only CM_128 continues towards clinical development. GSK-7975A inhibits SOCE induced by thapsigargin in isolated murine pancreatic acinar cells over the range 1-50 µM (IC50 ~3.4 µM) (24), inhibits endocytic vacuole formation (78) and reduces necrosis induced by toxins that cause acute pancreatitis (24, 78, 84). CM_128 is a new molecular entity, the effects of which we have recently confirmed to be similar(84). We have also shown that ORAI inhibition inhibits SOCE and necrosis in human pancreatic acinar cells, and found ORAI inhibition to reduce markedly the severity of multiple models of experimental acute pancreatitis.
Genetic knockout of the transient receptor potential canonical (TRPC) 3 channel (38), a non-selective cation channel regulated in part by STIM1 via TRPC1 (42), results in ~50% reduction of in vivo serum amylase elevation and edema formation induced by four injections of cerulein (38). These experiments supported a role for SOCE in acute pancreatitis, but in a single, mild model with few parameters of response.
As indicated we have defined the concentration-dependent inhibitory effects of GSK-7975A and CM_128 on SOCE and necrosis in murine and human pancreatic acinar cells induced by TLCS (60, 79) or CCK-8 (38, 63). The effects of CM_128 on ORAI1 were confirmed by examination of its effect on Ca2+ release-activated Ca2+ currents (ICRAC) (16, 24, 50), in ORAI1/STIM1-transfected HEK 293 cells (16). GSK-7975A was given at selected doses in vivo after induction of acute pancreatitis with TLCS (TLCS-AP) (40), seven injections of cerulein (CER-AP) (43) or ethanol and palmitoleic acid (FAEE-AP) (34). Since GSK-7975A markedly reduced all parameters of pathobiologic response in a dose-dependent manner, a high dose of GSK-7975A and separately CM_128 was begun at two different time points after disease induction to determine the effect of early versus late drug administration. Drug administration begun one hour after disease induction was highly effective in reducing parameters of the pathobiologic response (84), significantly more so than when begun six hours after disease induction, in all models. These data provide thorough preclinical validation for ORAI channel inhibition as a potential early treatment for acute pancreatitis.
We found GSK-7975A and the new molecular entity CM_128 to inhibit toxin-induced SOCE into murine and human pancreatic acinar cells in a concentration-dependent manner, exceeding >90% block of relative control values in some protocols (84). We also found both GSK-7975A and CM_128 to significantly reduce necrotic cell death pathway activation in murine and human pancreatic acinar cells exposed to TLCS, which induces acute pancreatitis in vivo (40, 43). While effects of GSK-7975A have been described on thapsigargin- and FAEE-induced murine pancreatic acinar SOCE (24), our studies found GSK-7975A to have a similarly critical effect on TLCS- and CCK-induced murine pancreatic acinar SOCE, as well as thapsigargin-induced human pancreatic acinar SOCE and TLCS-induced human pancreatic acinar necrotic cell death pathway activation (84). CM_128 showed higher potency (IC50 ~0.1 µM from ORAI1/STIM1-transfected HEK 293 cell patch clamp data), and unlike GSK-7975A, no loss of efficacy at high doses. Comprehensive in vivo evaluation using three diverse, clinically representative models of acute pancreatitis (84) with prior pharmacokinetic assessment demonstrated the validity of SOCE inhibition as a therapeutic approach. Thus administration of either compound within one hour following disease induction was markedly effective across a representative range of local and systemic biochemical, immunological and histological disease responses. These data provide robust confirmation of the hypothesis that cytosolic Ca2+ overload is a critical trigger of acute pancreatitis (83).
Further confirmation of the role of cytosolic Ca2+ overload in acute pancreatitis has come from our work with xanthines (35). We defined the inhibitory effects of methylxanthines on IP3R-mediated Ca2+ release from the pancreatic acinar ER store into the cytosol and potential application in acute pancreatitis. It has been shown that caffeine inhibits IP3Rs as well as IP3 production in a concentration dependent manner (75). We found that inhibition of IP3R-mediated Ca2+ release is attributable at least in part to an action on the IP3R, since xanthines inhibited IP3R-mediated Ca2+ release elicited by uncaged IP3 (35). Caffeine, theophylline and paraxanthine prevented physiological Ca2+ signaling and toxic elevations of [Ca2+]C induced by agents (CCK and TLCS) that cause acute pancreatitis, in a concentration dependent manner (500 µM to 10 mM), also inhibiting falls in mitochondrial membrane potential (∆ΨM) and necrotic cell death pathway activation. An inhibitory action on phosphodiesterase (PDE) preventing cAMP/cGMP degradation could not account for the effects on toxic [Ca2+]C overload, since additional cAMP/cGMP did not prevent these. Extending these findings in vivo, caffeine significantly reduced the severity of multiple, diverse models of acute pancreatitis (35). The combined concentrations of di- and trimethylxanthines after the 25 mg/kg caffeine protocol were within the range over which effects on both IP3R-mediated Ca2+ release and toxic elevations of [Ca2+]C were identified. Despite the half-life of caffeine in mice of ~60 min (4), the combined peak concentrations of di- and trimethylxanthines with 25 mg/kg caffeine regimen (7 injections) were >2 mM, and serum caffeine was >400 μM 6 h after last caffeine injection. Following similar protocols of 25 mg/kg theophylline or paraxanthine, concentrations were far below the effective range on IP3Rs but within the effective range on PDE (approaching 100 µM 10 min after the last dimethylxanthine injection) (20), and no protective effects on in vivo acute pancreatitis were seen. Since pancreatic cellular injury initiates and determines severity in acute pancreatitis, the protective effect of caffeine on acute pancreatitis is likely to have been mediated by inhibition of IP3R-mediated Ca2+ release.
3. The effects of calcium elevation in mitochondria:
The pancreatic acinar cell typifies non-excitable exocrine cells with a high secretory turnover that is heavily dependent on mitochondrial production of ATP (61). While zymogen activation has long been considered the principle mechanism of injury (41, 59), mitochondrial dysfunction has been increasingly implicated (12, 34, 44, 68, 70, 74, 80), presumed consequent upon intracellular Ca2+ overload induced by toxins that include bile acids and ethanol metabolites (5, 12, 34). Mitochondrial uptake of Ca2+ drives normal cellular bioenergetics, but high calcium loads induce increasingly drastic responses culminating in necrosis (30). Mitochondrial matrix Ca2+ overload leads to opening of the mitochondrial permeability transition pore (MPTP), a non-specific channel that forms in the inner mitochondrial membrane allowing passage of particles under 1500 Da, causing loss of ∆ψm essential to ATP production (30); recent evidence implicates F0F1 ATP synthase in MPTP formation (1, 27). MPTP opening is physiological in low conductance mode releasing Ca2+ and reactive oxygen species (ROS) to match metabolism with workload (17, 18), but pathological in high conductance mode compromising ATP production and inducing cell death (30); both functions are regulated by the mitochondrial matrix protein peptidyl-prolyl cis-trans isomerase (PPI, cyclophilin) D (also known as cyclophilin F) (76).
Our work has demonstrated that MPTP opening is critical to experimental acute pancreatitis, mediating impaired ATP production, defective autophagy, zymogen activation, inflammatory responses and necrosis(52), all features of acute pancreatitis at molecular, cellular and whole organism levels (59). We have established the general significance of MPTP opening as a central mechanism in the pathogenesis of acute pancreatitis, and the primary role in this process of Ca2+ overload. Patch clamp data show how tight control of cytosolic Ca2+ elevations essential to normal stimulus-secretion coupling by IP3Rs and ryanodine receptors (RyRs) (61) is lost in Wt but maintained in Ppif-/- pancreatic acinar cells, which lack functional MPTP, preserve ATP supply and clear Ca2+ more effectively. Coupling of ER IP3Rs and RyRs with outer mitochondrial membranes tightly localizes high Ca2+ concentrations (65), but may expose mitochondria to abnormal Ca2+ release, despite modulation by Bcl-2 family proteins (74). We have shown that pancreatitis toxins cause abnormal release of Ca2+ via IP3Rs and RyRs that overloads pancreatic acinar mitochondria (52), which are markedly sensitive to Ca2+ signals (58). The mitochondrial Ca2+ overload induces high conductance MPTP opening and dissipates ∆ψm, initiating collapse of ATP production, diminished Ca2+ clearance, PGAM5 (phosphoglycerate mutase family member 5, a mitochondrial protein phosphatase) activation and subsequent necrosis (52). Importantly for a disease without specific treatment, pharmacological MPTP inhibition (55, 84) administered after acute pancreatitis induction came close to preventing all injury, notably in the clinically relevant TLCS-AP.
For more than a century following an original postulate by Chiari (8), acute pancreatitis has been viewed as an autodigestive disease consequent on pathological zymogen activation (22, 29, 36, 41, 66). In experimental acute pancreatitis zymogens are activated inside acinar cells within minutes of toxin exposure (41, 47, 59, 63), which we have shown to result from induction of the MPTP, caused by and contributing to Ca2+ overload. Sustained Ca2+ overload may activate degradative calpains, phospholipases or other enzymes (76) and damage zymogen granules, inducing autophagic (47) and/or endolysosomal (71) responses that activate digestive enzymes. Such activation was not completely prevented by MPTP inhibition, however, this was likely from global cytosolic Ca2+ overload that was seen to be more effectively cleared in Ppif-/- cells, without which overload no enzyme activation occurs (63). Nevertheless, intracellular expression of trypsin per se without mitochondrial injury leads to apoptotic not necrotic pathway activation (22) and trypsinogen activation does not appear necessary for either local or systemic inflammation (13); knockout of cathepsin B greatly reduces trypsinogen activation with little effect on serum interleukin-6 or lung injury (29). Hereditary pancreatitis caused by cationic trypsinogen gene mutations rarely features clinically significant pancreatic necrosis (33, 85); further, systemic protease inhibition has had little success as a clinical strategy (59), suggesting that while zymogen activation contributes, it is not the critical driver of acute pancreatitis. Our work, however, shows that MPTP opening triggers defective autophagy, while inhibition of MPTP opening preserved ATP supply, increased the efficiency of autophagy and decreased zymogen activation. Together with major effects of MPTP opening on PGAM5 activation that implements necrosis (69, 82), and on local and systemic inflammatory responses, these findings now place mitochondrial injury centrally in acute pancreatitis.
Our data show that in pancreatic acinar cells IP3Rs and RyRs are vulnerable to specific toxins that markedly increase their Ca2+ channel open-state probabilities(52). Toxic transformation of Ca2+ channel function induced pancreatic acinar cell necrosis through Ca2+-dependent formation of the MPTP, with diminished ATP production the critical consequence. Toxic transformation by different toxins was specific to different second messengers, identifying potential for a variety of deleterious effects. ATP deficiency may be further exacerbated by fatty acids released on hydrolysis of FAEEs or triglycerides (56), which may inhibit beta-oxidation (12). Without sufficient ATP, cytosolic Ca2+ overload produces a vicious circle in which high affinity, low capacity SERCA and PMCA pump clearance of cytosolic Ca2+ is impaired, further mitochondrial injury sustained, and necrotic cell death accelerated (5, 12). Although the toxicity of cytosolic Ca2+ overload depends on Ca2+ store refilling from outside the cell (38, 63), specific second messenger receptor blockade demonstrated Ca2+ overload to be due completely to release from their Ca2+ channels (52), not direct effects of toxins on Ca2+ entry or extrusion.
Whereas the vast majority of previous studies undertaken to determine mechanisms and/or new targets in acute pancreatitis have used only one model, we have used four models (52) that are broadly representative of a range of etiologies, including biliary (TLCS-AP), hyperstimulation (CER-AP), ethanolic (FAEE-AP) and amino acid-induced (CDE-AP) (59, 72). Our findings in experimental acute pancreatitis are entirely consistent with those made in isolated mitochondria and cells, identifying a generalized mechanism of pancreatic injury and necrosis, confirmed in murine and human pancreatic acinar cells, pancreas lobules and tissue slices. Pancreatic necrosis drives the inflammasome (32), which can be induced by MPTP opening (54) and is part of the systemic inflammatory response contributing to multiple organ failure (62). Further pancreatic injury is driven through tumor necrosis factor receptor activation that also promotes MPTP opening (31) and calcium deregulation, activating calcineurin and calcineurin-dependent transcription factor nuclear factor of activated T cells (51).
4. Therapeutic avenues for acute pancreatitis:
Our novel human data support the potential applicability of SOCE inhibition as a treatment for clinical acute pancreatitis (Figure 2). Both GSK-7975A and CM_128 blocked SOCE promptly, shown to result in complete block of human ORAI1 by CM_128 (84). While an action on other ORAI channels cannot be excluded and could be desirable, ORAI1 is the primary channel for SOCE into pancreatic acinar cells (24, 45), blocked by both compounds. ORAI channels also contribute to inflammatory cell responses, including neutrophil migration and activation (3); inhibition of innate immune responses significantly reduces the severity of experimental acute pancreatitis (28), thus there may be a contribution from ORAI inhibition of immune cells. Nevertheless while knockout of ORAI1/STIM1 SOCE inhibits neutrophil functions, it does not prevent all (3), so the primary contribution of ORAI blockade in our experiments is likely to have been in the pancreas. Further, since SOCE inhibition for clinical acute pancreatitis would necessarily be short-term, inhibition of the adaptive immune system (3) would also be short-term. ORAI blockade has less effect on other cell types in which ORAI channels have a less prominent role, such as electrically excitable cells in which other ion channels, e.g. non-selective cation channels, have a larger role in Ca2+ entry (9). Non-selective cation channels, however, permit limited SOCE into pancreatic acinar cells (24, 38) that could sustain essential Ca2+ entry (9). Without such Ca2+ entry, continued activation of the plasma membrane Ca2+-ATPase pump upon secretagogue- or toxin-mediated release of Ca2+ from intracellular stores could deplete these stores to deleterious levels, inducing or exacerbating ER stress (48).
Both ORAI inhibitory compounds were administered after disease induction to model treatment of clinical acute pancreatitis, but delay in administration of either compound to six hours after disease induction resulted in diminished efficacy, dependent on the endpoint measured and the model employed (84). While biological time courses including that of acute pancreatitis are longer in humans than mice (15, 43, 59, 62), with pancreatic necrosis typically detected within days rather than hours (73), human pancreatic acinar necrotic cell death pathway activation may begin in clinical acute pancreatitis at an early stage after disease onset, shown here in mouse models within six hours of onset.
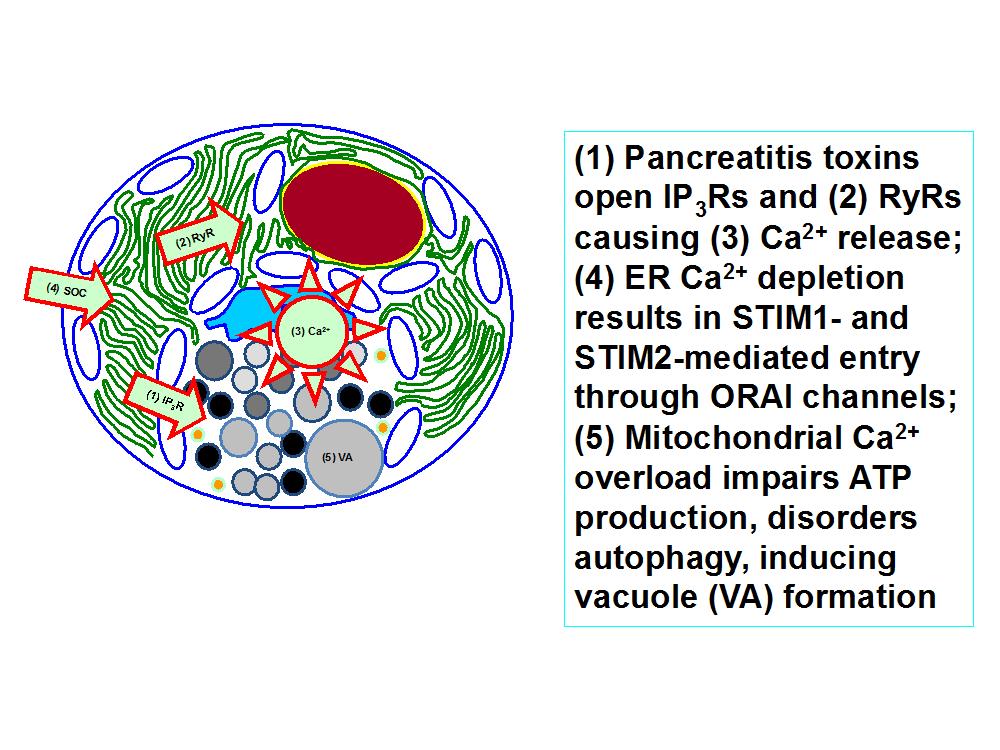
Figure 2. Abnormal Ca2+ signaling in the pancreatic acinar cell initiated by pancreatitis toxins (e.g. bile acids, FAEEs, hyperstimulation) causes injury dependent on continued store-operated Ca2+ (SOC) entry via ORAI channels. . As a consequence mitochondria are overloaded with Ca2+, failing to produce adequate ATP to clear the Ca2+ and protect the cell. Autophagy is defective and vacuoles develop presaging death of the cell. Ca2+ entry blockade prevents these events, avoiding prolonged, global cytosolic Ca2+ overload. This was first demonstrated by removal of Ca2+ from the external medium surrounding isolated cells and has since been shown using ORAI blockers applied to isolated human and murine pancreatic acinar cells. These findings have since been extended into three murine models of experimental acute pancreatitis, in which this strategy of Ca2+ entry inhibition with ORAI blockers has been demonstrated to be highly effective.
Door-to-needle times of less than 60 minutes are established guidelines for patients with acute myocardial infarction (30 min) (57) and acute ischemic stroke (60 min) (37), making every second count, with national and international quality improvement initiatives underway towards fully achieving these (19).
Although pancreatic necrosis has a less rapid time course and is characteristically not the result of major arterial occlusion (59), the translational implication of our work is that door-to-needle time is an important issue in administration of any treatment for acute pancreatitis that targets the pathogenesis of pancreatic injury, which drives the disease. Previously clinical trials of treatments for acute pancreatitis have ‘enriched’ recruitment with patients predicted to have severe disease (often with recruitment up to 72 h after admission) (77), which delays initiation of therapy. Furthermore, the expansion of disease categories from the original Atlanta Classification (mild and severe) (6) into the revised Atlanta (mild, moderate and severe) (2) and Determinants-Based (mild, moderate, severe, critical) (14) classification, further complicates patient selection from among these potentially overlapping sub-groups. To minimize door-to-needle time a quicker and more accurate approach to the selection of patients is required for trials of any therapy, such as that offered here with ORAI inhibition by CM_128, a novel molecular entity currently undergoing preclinical toxicological evaluation prior to phase I trials.
With respect to inhibition of Ca2+ release within the pancreatic acinar cell rather than Ca2+ entry into the cell, it is important to note that in our studies high doses of caffeine were required to reduce the severity of experimental acute pancreatitis. The most effective 25 mg/kg regimen extended into toxicity, indicative of a very narrow therapeutic index. At this dose the number of hourly injections had to be reduced from seven to two in FAEE-AP to avoid mortality; in CER-AP, 50 mg/kg resulted in caffeine intoxication syndrome, although at 25 mg/kg no visible side effects were observed. In humans even 10 mg/kg caffeine would be likely to induce caffeine intoxication, with florid neuro-excitotoxic and other undesirable side effects (20). There is marked individual variability in caffeine metabolism and pharmacokinetics (20);since the half-life in humans typically ranges from three to seven hours, repeated high doses would be hazardous, unless rapid therapeutic monitoring were to be possible. Nevertheless our study has demonstrated proof of principle that caffeine causes marked amelioration of experimental acute pancreatitis, largely through inhibition of IP3R-mediated signalling. Medicinal chemistry starting with the template of caffeine and/or other compounds that inhibit IP3R-mediated signalling could lead to more potent, selective and safer drug candidates for acute pancreatitis. This approach, however, might have effects on IP3R-mediated signalling in other cells, tissues and organs including the brain and other solid organs.
Our data from the cyclophilin D knockout and pharmacotherapy with Debio-025 (non-immunosuppressive derivative of cyclosporin A) or TRO40303 (in clinical development for other indications) show the potential for MPTP inhibition as an alternative strategy to ORAI inhibition (Figure 3).
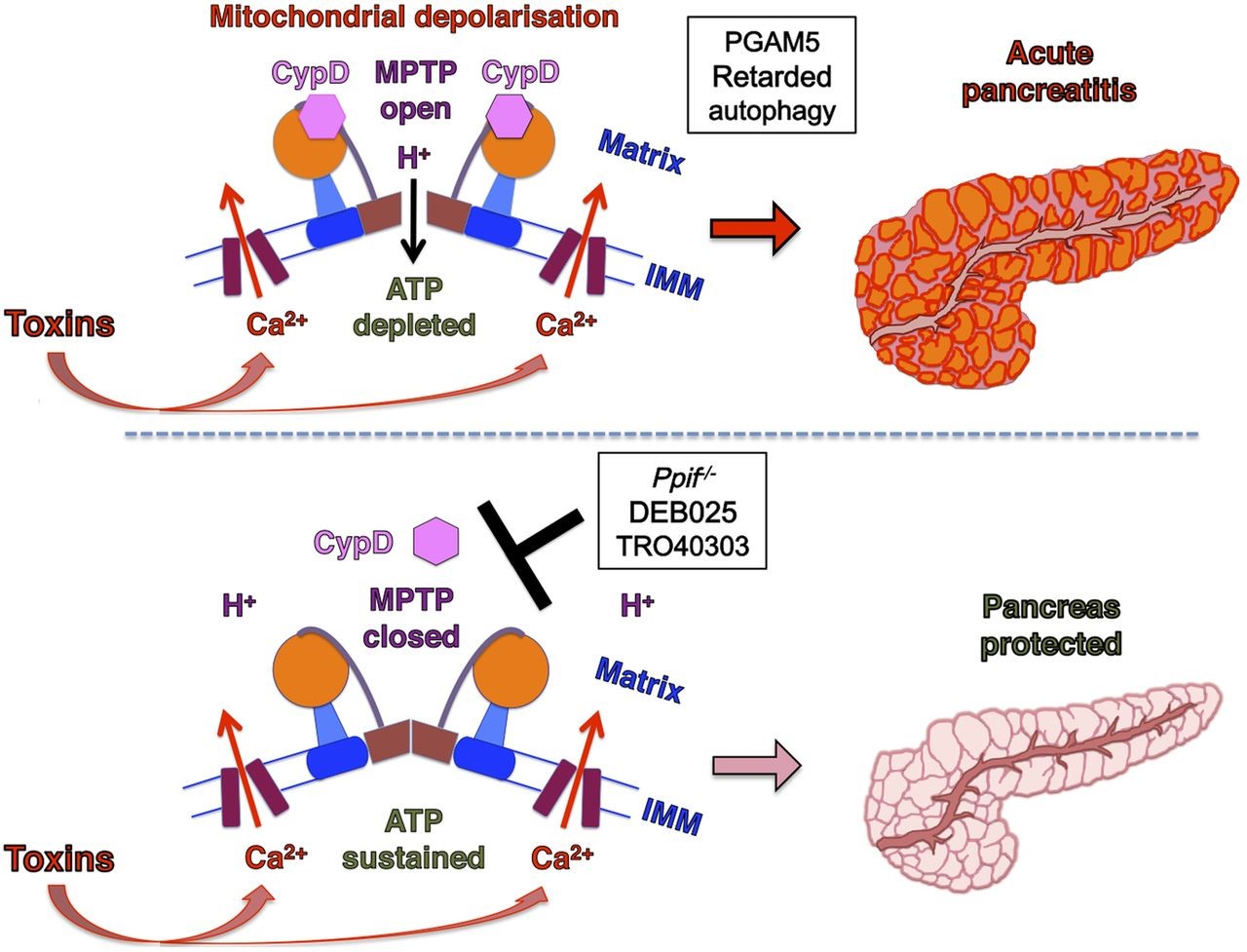
Figure 3. The MPTP plays a critical role in the development of acute pancreatitis. Pancreatitis toxins induce a sustained rise in [Ca2+]C that crosses the inner mitochondrial membrane (IMM) via the mitochondrial uniporter to enter the mitochondrial matrix. Consequent Ca2+-cyclophilin D (CypD) activation promotes MPTP opening, causing mitochondrial depolarization and impaired ATP production, failure of Ca2+ clearance and cell injury. When MPTP opening is inhibited by genetic (Ppif−/−) or pharmacological means (DEB025 or TR040303), mitochondrial membrane potential is preserved and ATP production sustained. This maintains cellular integrity to clear calcium more effectively and prevents the development of acute pancreatitis (lower panel) (Copyright © BMJ Publishing Group Ltd and British Society of Gastroenterology. All rights reserved: http://gut.bmj.com/content/65/8/1333.full).
The effect of this approach was remarkably effective in both isolated human and murine pancreatic acinar cells as well as in four models of experimental acute pancreatitis (52). The attractions of this approach are made stronger by the relative modest phenotype of the cyclophilin D knockout that is able to grow, develop and breed normally; there is an extensive range of pathologies that cyclophilin D knockout protects against, although capacity for exercise is reduced and there is some impact on memory in later life (17, 18). These effects are unlikely to be important in short-term administration as would be required in acute pancreatitis, and has not proven a problem in long-term administration of cyclosporin A.
Acknowledgements:
The findings described in this review are in part derived from three recent original open access articles (35, 52: Copyright © 2015 BMJ Publishing Group Ltd and British Society of Gastroenterology. All rights reserved. 85: Copyright © 2015 AGA Institute and Elsevier Inc. All rights reserved. See
35: http://gut.bmj.com/content/65/8/1333.full;
52: http://gut.bmj.com/content/early/2015/12/07/gutjnl-2015-309363.full
85: http://www.gastrojournal.org/article/S0016-5085(15)00571-5/fulltext).
We acknowledge funding support from Liverpool China Scholarship Council, CORE, the UK Medical Research Council, CalciMedica and the Biomedical Research Unit Funding scheme of the UK National Institute for Health Research. Robert Sutton is an NIHR Senior Investigator.
5. References:
- Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci U S A 111(29): 10580-10585, 2014. PMID: 24979777.
- Banks PA, Bollen TL, Dervenis C, Gooszen HG, Johnson CD, Sarr MG, et al. Classification of acute pancreatitis--2012: revision of the Atlanta classification and definitions by international consensus. Gut 62(1): 102-111, 2013. PMID: 23100216.
- Bergmeier W, Weidinger C, Zee I and Feske S. Emerging roles of store-operated Ca(2)(+) entry through STIM and ORAI proteins in immunity, hemostasis and cancer. Channels (Austin) 7(5): 379-391, 2013. PMID: 23511024.
- Bonati M, Latini R, Tognoni G, Young JF and Garattini S. Interspecies comparison of in vivo caffeine pharmacokinetics in man, monkey, rabbit, rat, and mouse. Drug Metab Rev 15(7): 1355-1383, 1984. PMID: 6543526.
- Booth DM, Murphy JA, Mukherjee R, Awais M, Neoptolemos JP, Gerasimenko OV, et al. Reactive oxygen species induced by bile acid induce apoptosis and protect against necrosis in pancreatic acinar cells. Gastroenterology 140(7): 2116-2125, 2011. PMID: 21354148.
- Bradley EL, 3rd. A clinically based classification system for acute pancreatitis. Summary of the International Symposium on Acute Pancreatitis, Atlanta, Ga, September 11 through 13, 1992. Arch Surg 128(5): 586-590, 1993. PMID: 8489394.
- Cancela JM, Van Coppenolle F, Galione A, Tepikin AV and Petersen OH. Transformation of local Ca2+ spikes to global Ca2+ transients: the combinatorial roles of multiple Ca2+ releasing messengers. EMBO J 21(5): 909-919, 2002. PMID: 11867519.
- Chiari H. Uber die Selbstverdauung des menschlichen Pankreas. Z Heilk 17: 69-96, 1896.
- Choi S, Maleth J, Jha A, Lee KP, Kim MS, So I, et al. The TRPCs-STIM1-Orai interaction. Handb Exp Pharmacol 223: 1035-1054, 2014. PMID: 24961979.
- Criddle DN, Booth DM, Mukherjee R, McLaughlin E, Green GM, Sutton R, et al. Cholecystokinin-58 and cholecystokinin-8 exhibit similar actions on calcium signaling, zymogen secretion, and cell fate in murine pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol 297(6): G1085-1092, 2009. PMID: 19815626.
- Criddle DN, McLaughlin E, Murphy JA, Petersen OH and Sutton R. The pancreas misled: signals to pancreatitis. Pancreatology 7(5-6): 436-446, 2007. PMID: 17898533.
- Criddle DN, Murphy J, Fistetto G, Barrow S, Tepikin AV, Neoptolemos JP, et al. Fatty acid ethyl esters cause pancreatic calcium toxicity via inositol trisphosphate receptors and loss of ATP synthesis. Gastroenterology 130(3): 781-793, 2006. PMID: 16530519.
- Dawra R, Sah RP, Dudeja V, Rishi L, Talukdar R, Garg P, et al. Intra-acinar trypsinogen activation mediates early stages of pancreatic injury but not inflammation in mice with acute pancreatitis. Gastroenterology 141(6): 2210-2217 e2212, 2011. PMID: 21875495.
- Dellinger EP, Forsmark CE, Layer P, Levy P, Maravi-Poma E, Petrov MS, et al. Determinant-based classification of acute pancreatitis severity: an international multidisciplinary consultation. Ann Surg 256(6): 875-880, 2012. PMID: 22735715.
- Demetrius L, Legendre S and Harremoes P. Evolutionary entropy: a predictor of body size, metabolic rate and maximal life span. Bull Math Biol 71(4): 800-818, 2009. PMID: 19172360.
- Derler I, Schindl R, Fritsch R, Heftberger P, Riedl MC, Begg M, et al. The action of selective CRAC channel blockers is affected by the Orai pore geometry. Cell Calcium 53(2): 139-151, 2013. PMID: 23218667.
- Elrod JW and Molkentin JD. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ J 77(5): 1111-1122, 2013. PMID: 23538482.
- Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA, et al. Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest 120(10): 3680-3687, 2010. PMID: 20890047.
- Fonarow GC, Zhao X, Smith EE, Saver JL, Reeves MJ, Bhatt DL, et al. Door-to-needle times for tissue plasminogen activator administration and clinical outcomes in acute ischemic stroke before and after a quality improvement initiative. JAMA 311(16): 1632-1640, 2014. PMID: 24756513.
- Fredholm BB, Battig K, Holmen J, Nehlig A and Zvartau EE. Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol Rev 51(1): 83-133, 1999. PMID: 10049999.
- Futatsugi A, Nakamura T, Yamada MK, Ebisui E, Nakamura K, Uchida K, et al. IP3 receptor types 2 and 3 mediate exocrine secretion underlying energy metabolism. Science 309(5744): 2232-2234, 2005. PMID: 16195467.
- Gaiser S, Daniluk J, Liu Y, Tsou L, Chu J, Lee W, et al. Intracellular activation of trypsinogen in transgenic mice induces acute but not chronic pancreatitis. Gut 60(10): 1379-1388, 2011. PMID: 21471572.
- Gerasimenko JV, Flowerdew SE, Voronina SG, Sukhomlin TK, Tepikin AV, Petersen OH, et al. Bile acids induce Ca2+ release from both the endoplasmic reticulum and acidic intracellular calcium stores through activation of inositol trisphosphate receptors and ryanodine receptors. J Biol Chem 281(52): 40154-40163, 2006. PMID: 17074764.
- Gerasimenko JV, Gryshchenko O, Ferdek PE, Stapleton E, Hebert TO, Bychkova S, et al. Ca2+ release-activated Ca2+ channel blockade as a potential tool in antipancreatitis therapy. Proc Natl Acad Sci U S A 110(32): 13186-13191, 2013. PMID: 23878235.
- Gerasimenko JV, Lur G, Ferdek P, Sherwood MW, Ebisui E, Tepikin AV, et al. Calmodulin protects against alcohol-induced pancreatic trypsinogen activation elicited via Ca2+ release through IP3 receptors. Proc Natl Acad Sci U S A 108(14): 5873-5878, 2011. PMID: 21436055.
- Gerasimenko JV, Lur G, Sherwood MW, Ebisui E, Tepikin AV, Mikoshiba K, et al. Pancreatic protease activation by alcohol metabolite depends on Ca2+ release via acid store IP3 receptors. Proc Natl Acad Sci U S A 106(26): 10758-10763, 2009. PMID: 19528657.
- Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A 110(15): 5887-5892, 2013. PMID: 23530243.
- Gukovskaya AS, Vaquero E, Zaninovic V, Gorelick FS, Lusis AJ, Brennan ML, et al. Neutrophils and NADPH oxidase mediate intrapancreatic trypsin activation in murine experimental acute pancreatitis. Gastroenterology 122(4): 974-984, 2002. PMID: 11910350.
- Halangk W, Lerch MM, Brandt-Nedelev B, Roth W, Ruthenbuerger M, Reinheckel T, et al. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J Clin Invest 106(6): 773-781, 2000. PMID: 10995788.
- Halestrap AP and Richardson AP. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J Mol Cell Cardiol 78: 129-141, 2015. PMID: 25179911.
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137(6): 1100-1111, 2009. PMID: 19524512.
- Hoque R, Sohail M, Malik A, Sarwar S, Luo Y, Shah A, et al. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology 141(1): 358-369, 2011. PMID: 21439959.
- Howes N, Lerch MM, Greenhalf W, Stocken DD, Ellis I, Simon P, et al. Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin Gastroenterol Hepatol 2(3): 252-261, 2004. PMID: 15017610.
- Huang W, Booth DM, Cane MC, Chvanov M, Javed MA, Elliott VL, et al. Fatty acid ethyl ester synthase inhibition ameliorates ethanol-induced Ca2+-dependent mitochondrial dysfunction and acute pancreatitis. Gut 63(8): 1313-1324, 2014. PMID: 24162590.
- Huang W, Cane MC, Mukherjee R, Szatmary P, Zhang X, Elliott V, et al. Caffeine protects against experimental acute pancreatitis by inhibition of inositol 1,4,5-trisphosphate receptor-mediated Ca2+ release. Gut, 2015. PMID: 26642860.
- Husain SZ, Prasad P, Grant WM, Kolodecik TR, Nathanson MH and Gorelick FS. The ryanodine receptor mediates early zymogen activation in pancreatitis. Proc Natl Acad Sci U S A 102(40): 14386-14391, 2005. PMID: 16186498.
- Jauch EC, Saver JL, Adams HP, Jr., Bruno A, Connors JJ, Demaerschalk BM, et al. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 44(3): 870-947, 2013. PMID: 23370205.
- Kim MS, Hong JH, Li Q, Shin DM, Abramowitz J, Birnbaumer L, et al. Deletion of TRPC3 in mice reduces store-operated Ca2+ influx and the severity of acute pancreatitis. Gastroenterology 137(4): 1509-1517, 2009. PMID: 19622358.
- Kruger B, Albrecht E and Lerch MM. The role of intracellular calcium signaling in premature protease activation and the onset of pancreatitis. Am J Pathol 157(1): 43-50, 2000. PMID: 10880374.
- Laukkarinen JM, Van Acker GJ, Weiss ER, Steer ML and Perides G. A mouse model of acute biliary pancreatitis induced by retrograde pancreatic duct infusion of Na-taurocholate. Gut 56(11): 1590-1598, 2007. PMID: 17591621.
- Leach SD, Modlin IM, Scheele GA and Gorelick FS. Intracellular activation of digestive zymogens in rat pancreatic acini. Stimulation by high doses of cholecystokinin. J Clin Invest 87(1): 362-366, 1991. PMID: 1985109.
- Lee KP, Choi S, Hong JH, Ahuja M, Graham S, Ma R, et al. Molecular determinants mediating gating of Transient Receptor Potential Canonical (TRPC) channels by stromal interaction molecule 1 (STIM1). J Biol Chem 289(10): 6372-6382, 2014. PMID: 24464579.
- Lerch MM and Gorelick FS. Models of acute and chronic pancreatitis. Gastroenterology 144(6): 1180-1193, 2013. PMID: 23622127.
- Lerch MM, Halangk W and Mayerle J. Preventing pancreatitis by protecting the mitochondrial permeability transition pore. Gastroenterology 144(2): 265-269, 2013. PMID: 23260493.
- Lur G, Haynes LP, Prior IA, Gerasimenko OV, Feske S, Petersen OH, et al. Ribosome-free terminals of rough ER allow formation of STIM1 puncta and segregation of STIM1 from IP(3) receptors. Curr Biol 19(19): 1648-1653, 2009. PMID: 19765991.
- Lur G, Sherwood MW, Ebisui E, Haynes L, Feske S, Sutton R, et al. InsP(3)receptors and Orai channels in pancreatic acinar cells: co-localization and its consequences. Biochem J 436(2): 231-239, 2011. PMID: 21568942.
- Mareninova OA, Hermann K, French SW, O'Konski MS, Pandol SJ, Webster P, et al. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J Clin Invest 119(11): 3340-3355, 2009. PMID: 19805911.
- Mekahli D, Bultynck G, Parys JB, De Smedt H and Missiaen L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb Perspect Biol 3(6), 2011. PMID: 21441595.
- Mooren F, Hlouschek V, Finkes T, Turi S, Weber IA, Singh J, et al. Early changes in pancreatic acinar cell calcium signaling after pancreatic duct obstruction. J Biol Chem 278(11): 9361-9369, 2003. PMID: 12522141.
- Muik M, Schindl R, Fahrner M and Romanin C. Ca(2+) release-activated Ca(2+) (CRAC) current, structure, and function. Cell Mol Life Sci 69(24): 4163-4176, 2012. PMID: 22802126.
- Muili KA, Wang D, Orabi AI, Sarwar S, Luo Y, Javed TA, et al. Bile acids induce pancreatic acinar cell injury and pancreatitis by activating calcineurin. J Biol Chem 288(1): 570-580, 2013. PMID: 23148215.
- Mukherjee R, Mareninova OA, Odinokova IV, Huang W, Murphy J, Chvanov M, et al. Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: inhibition prevents acute pancreatitis by protecting production of ATP. Gut 65(8): 1333-1346, 2016. PMID: 26071131.
- Murphy JA, Criddle DN, Sherwood M, Chvanov M, Mukherjee R, McLaughlin E, et al. Direct activation of cytosolic Ca2+ signaling and enzyme secretion by cholecystokinin in human pancreatic acinar cells. Gastroenterology 135(2): 632-641, 2008. PMID: 18555802.
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12(3): 222-230, 2011. PMID: 21151103.
- Naoumov NV. Cyclophilin inhibition as potential therapy for liver diseases. J Hepatol 61(5): 1166-1174, 2014. PMID: 25048953.
- Navina S, Acharya C, DeLany JP, Orlichenko LS, Baty CJ, Shiva SS, et al. Lipotoxicity causes multisystem organ failure and exacerbates acute pancreatitis in obesity. Sci Transl Med 3(107): 107ra110, 2011. PMID: 22049070.
- O'Gara PT, Kushner FG, Ascheim DD, Casey DE, Jr., Chung MK, de Lemos JA, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 127(4): e362-425, 2013. PMID: 23247304.
- Odinokova IV, Sung KF, Mareninova OA, Hermann K, Evtodienko Y, Andreyev A, et al. Mechanisms regulating cytochrome c release in pancreatic mitochondria. Gut 58(3): 431-442, 2009. PMID: 18596195.
- Pandol SJ, Saluja AK, Imrie CW and Banks PA. Acute pancreatitis: bench to the bedside. Gastroenterology 132(3): 1127-1151, 2007. PMID: 17383433.
- Petersen OH and Sutton R. Ca2+ signalling and pancreatitis: effects of alcohol, bile and coffee. Trends Pharmacol Sci 27(2): 113-120, 2006. PMID: 16406087.
- Petersen OH and Tepikin AV. Polarized calcium signaling in exocrine gland cells. Annu Rev Physiol 70: 273-299, 2008. PMID: 17850212.
- Petrov MS, Shanbhag S, Chakraborty M, Phillips AR and Windsor JA. Organ failure and infection of pancreatic necrosis as determinants of mortality in patients with acute pancreatitis. Gastroenterology 139(3): 813-820, 2010. PMID: 20540942.
- Raraty M, Ward J, Erdemli G, Vaillant C, Neoptolemos JP, Sutton R, et al. Calcium-dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc Natl Acad Sci U S A 97(24): 13126-13131, 2000. PMID: 11087863.
- Rice LV, Bax HJ, Russell LJ, Barrett VJ, Walton SE, Deakin AM, et al. Characterization of selective Calcium-Release Activated Calcium channel blockers in mast cells and T-cells from human, rat, mouse and guinea-pig preparations. Eur J Pharmacol 704(1-3): 49-57, 2013. PMID: 23454522.
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280(5370): 1763-1766, 1998. PMID: 9624056.
- Saluja A, Saluja M, Villa A, Leli U, Rutledge P, Meldolesi J, et al. Pancreatic duct obstruction in rabbits causes digestive zymogen and lysosomal enzyme colocalization. J Clin Invest 84(4): 1260-1266, 1989. PMID: 2477393.
- Saluja AK, Bhagat L, Lee HS, Bhatia M, Frossard JL and Steer ML. Secretagogue-induced digestive enzyme activation and cell injury in rat pancreatic acini. Am J Physiol 276(4 Pt 1): G835-842, 1999. PMID: 10198325.
- Schild L, Matthias R, Stanarius A, Wolf G, Augustin W and Halangk W. Induction of permeability transition in pancreatic mitochondria by cerulein in rats. Mol Cell Biochem 195(1-2): 191-197, 1999. PMID: 10395083.
- Sekine S, Kanamaru Y, Koike M, Nishihara A, Okada M, Kinoshita H, et al. Rhomboid protease PARL mediates the mitochondrial membrane potential loss-induced cleavage of PGAM5. J Biol Chem 287(41): 34635-34645, 2012. PMID: 22915595.
- Shalbueva N, Mareninova OA, Gerloff A, Yuan J, Waldron RT, Pandol SJ, et al. Effects of oxidative alcohol metabolism on the mitochondrial permeability transition pore and necrosis in a mouse model of alcoholic pancreatitis. Gastroenterology 144(2): 437-446 e436, 2013. PMID: 23103769.
- Sherwood MW, Prior IA, Voronina SG, Barrow SL, Woodsmith JD, Gerasimenko OV, et al. Activation of trypsinogen in large endocytic vacuoles of pancreatic acinar cells. Proc Natl Acad Sci U S A 104(13): 5674-5679, 2007. PMID: 17363470.
- Simon P, Weiss FU, Zimmer KP, Koch HG and Lerch MM. Acute and chronic pancreatitis in patients with inborn errors of metabolism. Pancreatology 1(5): 448-456, 2001. PMID: 12120223.
- Spanier BW, Nio Y, van der Hulst RW, Tuynman HA, Dijkgraaf MG and Bruno MJ. Practice and yield of early CT scan in acute pancreatitis: a Dutch Observational Multicenter Study. Pancreatology 10(2-3): 222-228, 2010. PMID: 20484959.
- Sung KF, Odinokova IV, Mareninova OA, Rakonczay Z, Jr., Hegyi P, Pandol SJ, et al. Prosurvival Bcl-2 proteins stabilize pancreatic mitochondria and protect against necrosis in experimental pancreatitis. Exp Cell Res 315(11): 1975-1989, 2009. PMID: 19331832.
- Toescu EC, O'Neill SC, Petersen OH and Eisner DA. Caffeine inhibits the agonist-evoked cytosolic Ca2+ signal in mouse pancreatic acinar cells by blocking inositol trisphosphate production. J Biol Chem 267(33): 23467-23470, 1992. PMID: 1429689.
- Vandenabeele P, Galluzzi L, Vanden Berghe T and Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11(10): 700-714, 2010. PMID: 20823910.
- Villatoro E, Mulla M and Larvin M. Antibiotic therapy for prophylaxis against infection of pancreatic necrosis in acute pancreatitis. Cochrane Database Syst Rev(5): CD002941, 2010. PMID: 20464721.
- Voronina S, Collier D, Chvanov M, Middlehurst B, Beckett AJ, Prior IA, et al. The role of Ca2+ influx in endocytic vacuole formation in pancreatic acinar cells. Biochem J 465(3): 405-412, 2015. PMID: 25370603.
- Voronina S, Longbottom R, Sutton R, Petersen OH and Tepikin A. Bile acids induce calcium signals in mouse pancreatic acinar cells: implications for bile-induced pancreatic pathology. J Physiol 540(Pt 1): 49-55, 2002. PMID: 11927668.
- Voronina SG, Barrow SL, Simpson AW, Gerasimenko OV, da Silva Xavier G, Rutter GA, et al. Dynamic changes in cytosolic and mitochondrial ATP levels in pancreatic acinar cells. Gastroenterology 138(5): 1976-1987, 2010. PMID: 20102715.
- Voronina SG, Gryshchenko OV, Gerasimenko OV, Green AK, Petersen OH and Tepikin AV. Bile acids induce a cationic current, depolarizing pancreatic acinar cells and increasing the intracellular Na+ concentration. J Biol Chem 280(3): 1764-1770, 2005. PMID: 15536077.
- Wang Z, Jiang H, Chen S, Du F and Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148(1-2): 228-243, 2012. PMID: 22265414.
- Ward JB, Petersen OH, Jenkins SA and Sutton R. Is an elevated concentration of acinar cytosolic free ionised calcium the trigger for acute pancreatitis? Lancet 346(8981): 1016-1019, 1995. PMID: 7475553.
- Wen L, Voronina S, Javed MA, Awais M, Szatmary P, Latawiec D, et al. Inhibitors of ORAI1 Prevent Cytosolic Calcium-Associated Injury of Human Pancreatic Acinar Cells and Acute Pancreatitis in 3 Mouse Models. Gastroenterology 149(2): 481-492 e487, 2015. PMID: 25917787.
- Whitcomb DC, Gorry MC, Preston RA, Furey W, Sossenheimer MJ, Ulrich CD, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet 14(2): 141-145, 1996. PMID: 8841182.