Methods Type:
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2016.33
| Attachment | Size |
|---|---|
| 497.11 KB |
1. Introduction
Acute pancreatitis is a serious gastroenterological disease characterized by inflammation and damage of the exocrine pancreas. It is generally self-limiting, but in up to 20% of the cases severe clinical complications develop resulting in severe acute pancreatitis with a resulting mortality rate of up to 30% (25, 39, 40). Severe acute pancreatitis is associated with systemic inflammation, multi-organ failure, and high mortality. Unfortunately, there is currently no specific pharmacotherapy to treat or prevent the progression of this disease.
The most common etiological factors associated with acute pancreatitis in adults are gallstones and alcohol abuse, although in 20-30% of cases the cause is not known (6). In contrast, common etiologic factors in children include drugs, infections, trauma, and anatomical abnormalities (32). Interestingly, in a review of the literature Benifla and Weizman reported that approximately 10% of pediatric cases of acute pancreatitis resulted from viral infections (3).
A number of viruses are known to replicate in the pancreas and cause pancreatitis; among these are the coxsackieviruses (26). Coxsackieviruses are enteroviruses that are most commonly associated with myocarditis in humans. Coxsackieviruses are divided into two groups A and B, consisting of 24 and 6 serotypes respectively. The group B coxsackieviruses are common human pathogens. Although typically associated with heart disease, these viruses have been isolated from the pancreata of patients with pancreatitis (11, 26, 46). Furthermore, coxsackieviruses have been shown to replicate to high titers in the pancreas and cause extensive pancreatic tissue damage in experimental animal models (30, 34). In fact, in mice, if these viruses do not replicate to high titer in the pancreas, they do not cause heart disease (13, 34). Also, outbreaks of coxsackievirus-associated infections have been associated with increased cases of pancreatitis (20, 21). Although viral infection is a more common cause of acute pancreatitis in children, individual case studies have established coxsackievirus as a cause of acute pancreatitis in adults (7, 9, 16). Additionally, several studies have detected elevated antibody titers against coxsackievirus among patients with acute pancreatitis (2, 4, 23). Over a five-year period, Ozsvar et al. observed elevated antibody titers against coxsackieviruses in 34% of patients presenting with acute, recurrent, or chronic pancreatitis (23). Thus, it is evident that coxsackievirus infection of humans can cause acute pancreatitis.
Regardless of the etiology of acute pancreatitis, the seminal event responsible for initiation of the disease is thought to be common to all, namely the inappropriate activation of pancreatic digestive enzymes and autodigestion of the organ (36). A number of preclinical animal models of acute pancreatitis have been established. These include hyperstimulation of the pancreas with the cholecystokinin ortholog, caerulein, bile duct ligation, infusion of bile salts into the pancreatic duct, altered diets, and administration of basic amino acids (17). Although a tremendous amount of information regarding the molecular events that result in acute pancreatitis has been gained using these models, none of these insults to the pancreas occur in humans. Therefore, the physiologic relevance of these models to acute pancreatitis in humans is not clear. Because of this, one of the limitations in the study of pancreatic injury and acute pancreatitis in humans is the paucity of animal models that are biologically relevant to acute pancreatitis in human beings. Using group B serotype 3 coxsackieviruses (CVB3) isolated from individuals that have suffered from CVB3 infections, we have established a model of acute pancreatitis that is physiologically relevant to the disease in humans.
2. Materials and Reagents
2.1 Materials
- Agarose
- Powdered Dulbecco’s Modified Eagle’s Media
- Fetal Bovine Serum
- Bechman SW 28 or SW 41 rotor and tubes
- Formaldehyde
- Crystal violet
- Glass homogenizers (Dounce Tissue Grinders)
- 12 well tissue culture plates
2.2 Reagents
a. Preparation of 0.5% Agarose/DMEM
The 0.5% agarose/DMEM solution is made by preparing 2 separate solutions: a 1% solution of agarose and a solution of 2X DMEM. The 1% agarose solution is prepared by adding 1 gram of agarose/100 mL of distilled water and sterilized by autoclaving. The agarose solution is then cooled in a water bath to 55 oC. The 2X DMEM solution is prepared by adding twice the recommended weight of powdered DMEM and sodium bicarbonate to the volume you wish to prepare (normally 100 mL). The pH is adjusted to 7.2, it is filter sterilized through a 0.22 μm filter, and warmed to 55 oC. The 0.5% agarose/DMEM solution is prepared by combining equal volumes of the agarose and 2X DMEM solutions immediately before using it to overlaying the cells.
b. Preparation of 20 % formalin
Formalin is a stock solution of 37% formaldehyde. To prepare 20% formalin the stock formalin solution is diluted 1:5 using distilled water resulting in 7.4% formaldehyde
c. Preparation of 1% crystal violet
Crystal violet is prepared by adding 5 g of crystal violet to 395 mL of H2O and 105 mL of 20% ethanol. The solution is then filtered through a 0.22 μm filter.
3. Methods
CVB3 viruses are human pathogens (26, 34). Because of this, when working with CVB3, BSL2 precautions must be used. All work with these viruses should be performed in an approved type II biosafety cabinet, and any potentially contaminated equipment or solid waste must be decontaminated with a 10% bleach solution or by autoclaving. After working with tissues or solutions that may be contaminated with CVB3, all surfaces must be cleaned with a 10% bleach solution.
The CVB3 strains that we use are human pathogens, which have been isolated from individuals that have suffered CVB3 infections. These viruses have not been adapted to mice but cause disease in mice that is similar to that observed in humans (34). Because of this, we cultivate the virus in cell lines of human origin, normally HeLa cells.
a. Viral Growth and Isolation
- HeLa cells are seeded at 5 x 106 cells/75 cm2 flask (a concentration that will yield approximately 80% confluence) and incubated overnight.
- The following day, the growth media (DMEM containing 10% FBS and antibiotics) is removed by careful aspiration so as not to disturb the cell sheet. The cells are infected with the virus at a multiplicity of infection (MOI) of 0.01-0.001 in a 5 mL volume of DMEM containing 2% FBS. For example, we infect these cultures with 5 x 104 plaque forming units (PFU). This low MOI is used to limit the production of defective interfering particles.
- The infected cells are incubated at 37 oC for 30 minutes with occasional shaking.
- The viral inoculum is removed by aspiration and replaced with growth medium. The flasks are then incubated for 2-3 days at 37 oC, until the cell sheet is destroyed.
- The flasks are subjected to 3 cycles of freeze/thaw at -70 oC, and the virus containing supernatant is collected.
- The cellular debris is removed by centrifugation in a SW28 rotor at 25,000 rpm at 8 oC for 30 minutes or in a SW41 rotor at 25,000 rpm at 8 oC for 20 minutes. The clarified virus containing supernatant is collected and stored at -70 oC. If components present in the growth medium are not a concern in experiments this clarified supernatant can be used.
- If virus devoid of growth medium components is needed, the virus can be further purified by centrifugation through sucrose. Using SW28 tubes, add 12 mL of 30% sucrose, prepared in 1 M NaCl, 50 mM Tris (pH of 7.5). The culture supernatant is carefully dripped down the side of the tube so that it forms a layer on top of the sucrose. In a similar fashion, 4 drops of glycerol are dripped into the tube. The samples are centrifuged at 25,000 rpm overnight at 8 oC.
- The supernatant is removed, being careful not to disrupt the loose and barely visible pellet. The virus is suspended in 0.1 M NaCl by gently rocking the pellet. The purified virus is stored at -70 oC.
- The virus is titrated as described below. Note: coxsackievirus B preparations degrade at -20 oC by a titratable amount in one week; thus, long-term storage needs to be at -70 oC.
b. Viral Titration by Plaque Assay
We use HeLa cells to grow and titrate CVB3.
- If titrating virus from tissues, the tissue is weighed and transferred to a glass homogenizer (Dounce Tissue Grinders).
- A 10X volume of DMEM containing 2% fetal bovine serum (FBS) is added and the tissue is homogenized by 20-30 strokes. For example, 1 mL of DMEM is added to a tissue sample weighing 100 mg. This is effectively a 1:10 dilution of the tissue sample and must be considered when calculating the final viral titer.
- Prepare 10-fold dilutions of the tissue homogenate or virus containing solution (100 microliters into 900 microliters) in DMEM containing 2% FBS.
- 12 well plates are seeded at a density of 2 X 105 HeLa cells/well and incubated overnight. This will result in the wells being approximately 80% confluent the next morning.
- Each dilution of interest is assayed in triplicate (three wells) by replacing the growth media with 250 microliters of the appropriate dilution.
- The plates are incubated at 37 oC for 1 hour, with gentle rocking every 15 minutes.
- The virus containing solution is removed, and each well is carefully overlaid with 1 mL of 0.5% agarose/DMEM, making sure not to pipette the solution directly onto the cell sheet.
- The agarose is allowed to solidify at room temperature, and the plates are transferred to an incubator and incubated at 37 oC for 2 days.
- After incubation, the samples are fixed by adding 1 mL of 20% formalin to each well and incubating at room temperature for 15 minutes. This inactivates the virus.
- The formalin is removed from the wells, and the agarose overlay is removed from the wells with a spatula, being careful not to scratch the cell sheet.
- The fixed cells are then stained with 0.5 mL of 1% crystal violet for 1-2 minutes.
- The crystal violet is removed and the wells are washed 2-3 times with H2O. Plaques are visible as clear areas in the cell sheet (Figure 1). Most likely, one or more of the wells will contain too many plaques to accurately count or no plaques at all. The titer is expressed as PFU/gram of tissue or PFU/mL (depending on the starting material).
- The viral titer is determined by counting the number of plaques on three wells of a given dilution. The number of plaques counted is divided by three (number of wells/dilution) and then multiplied by 4 (because only 0.25 mL of viral solution is added to each well). This will yield PFU/g or PFU/mL.
- Example: A plate containing virus diluted to 10-6, 10-7, 10-8, 10-9 is prepared. On the three 10-7 wells, 4, 5, and 6 plaques are counted for a total of 15 plaques (Figure 1).
Calculations: (15 plaques)/(3 wells)*4 =20 PFU.
Because the plaques were counted on the 10-7 dilution the viral titer is 20 X 107 or 2.0 X 108 PFU/mL.
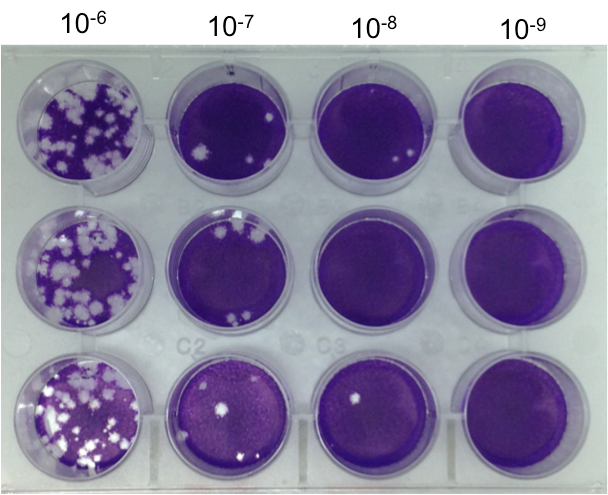
Figure 1. Plaque Assay. Representative Plaque Assay Plate. Wells were seeded with HeLa cells at a density of 2 X 105/well and incubated overnight. The following day the cells were infected and overlaid with 0.5%agarose/DMEM as described in the text. The plates were incubated for two days, the cells were fixed, the agarose overlays were removed and the cell sheet was stained with crystal violet. There were 15 plaques detected at the 10-7 dilution therefore the titer of the viral sample was 2 X 108/ml.
c. Viral Infection and Generation of Pancreatitis
It has been shown that CVB3 replication, as well as the pathogenesis induced by CVB3 infection in the heart of mice is dependent upon the strain of virus and the strain of mice used in the study (12, 42). It is important to note that the virulence and, perhaps, the course of the disease depend upon the specific strain of virus and the genetic make up of the mice, some virus strains being avirulent and some mouse strains being more resistant to the virus than others (14, 34). Because of this, the viral inoculum will vary depending on the strain of mice and virus used. If a virulent virus strain is used in a susceptible strain of mice all animals will develop pancreatitis. CVB3/CO and CVB3/28 cause overt pancreatic damage in C57BL/6 or Balb/c mice. In contrast, CVB3/GA infects the pancreas but does not cause histological damage (33, 34). Mice are maintained on standard lab chow and water ad libitum unless experimental procedures require otherwise. We normally inoculate mice by intraperitoneal (IP) injection of 0.2 mL of virus at a viral titer of 2.5 X 106 PFU/mL (5 x 105 PFU/mouse) in a type II safety cabinet. Because CVB3 is a human pathogen, as well as a mouse pathogen infected animals can infect other mice or laboratory personnel. Therefore, after infection, mice are maintained in a designated ABL2 facility. The normal transmission of CVB3 is oral fecal, so food and bedding may be contaminated and must be treated as such.
Histological analysis of pancreatic tissue from mice infected with CVB3 demonstrate extensive inflammation of the exocrine pancreas and acinar cell necrosis in accordance with a classification of moderate acute pancreatitis in humans, high morbidity without mortality. The inflammatory infiltrate is characterized primarily by monocytes (T-cells, B-cells, and natural killer cells) (27, 28). Although some strains of coxsackievirus have been shown to cause damage to the islets, the vast majority of CVB strains cause extensive damage to the acinar cells but spare the islets and the ducts (19, 45, 46),(37). Although there is some controversy regarding the mechanism by which CVB causes pancreatic tissue damage it appears that the damage observed in the pancreata of mice infected with coxsackievirus is the result of virus-induced cytolysis, autodigestion by pancreatic enzymes, and inflammatory cell-mediated tissue damage (10, 19, 27, 28).
In experimentally infected mice, the viral titers in the spleen peak 2-4 days post infection (PI) (30). Because of the highly vascular nature of the spleen, the titer in the spleen presumably represents the titer in the blood (level of viremia) of infected animals. Serum amylase and lipase levels peak 4 days PI after which they return to near normal levels (30). The peak in pancreatic tissue damage occurs approximately 6 days PI. Although the majority of the pancreas is affected, unaffected areas can be detected. The affected areas are characterized by extensive necrosis, edema, and infiltration of inflammatory cells (Figure 2C). Following the peak in damage, the affected areas of the exocrine pancreas demonstrate signs of repair with the reappearance of discernable pancreatic architecture and organization of acinar cells into acini. This repair process is apparent by 8 days PI (Figure 2D). By twelve days post infection, many areas appear normal. We have observed the accumulation of intrapancreatic fat during the regenerative process of C57BL/6 mice infected with CVB3/CO (Figure 2F). This fat appears to persist and accumulate over time. Interestingly, this accumulation of fat in the pancreas is attenuated in other strains of mice and total regeneration of the damaged pancreas is observed (45). Thus, it appears the regenerative response to CVB3 infection in mice is dependent on the strain of mice and perhaps the characteristics of the specific virus strain.
4. Discussion
We have established a physiologically relevant preclinical model of acute pancreatitis. This model utilizes coxsackieviruses isolated from humans who have suffered from CVB3 infection. Importantly, these viruses are not humanized and recapitulate many of the histologic changes observed in human acute pancreatitis (34).
In experimentally infected mice, the highest viral titer in the spleen is observed 2-4 days post infection (PI). Because of the highly vascular nature of the spleen, the titer in the spleen presumably represents the titer in the blood (level of viremia) of infected animals. The peak in pancreatic tissue damage occurs approximately 6 days PI. Although the majority of the pancreas is affected, unaffected areas can be detected. The affected areas are characterized by extensive necrosis, edema, and infiltration of inflammatory cells (Figure 2C). Following the peak in damage, the affected areas of the exocrine pancreas demonstrated signs of repair with the reappearance of discernable pancreatic architecture and organization of acinar cells into acini. This repair process is apparent by 8 days PI. By twelve days post infection, many areas appear normal. We have observed the accumulation of intrapancreatic fat during the regenerative process of C57BL/6 mice infected with CVB3/CO (Figure 2F). This fat appears to persist and accumulate over time. Interestingly, this accumulation of fat in the pancreas is attenuated in other strains of mice. Thus, it appears the regenerative response to CVB3 infection in mice is dependent on the strain of mice and the characteristics of the specific virus strain. Interestingly, infection of humans with mumps virus and mice with reovirus 1 and 2 have also been shown to cause fatty replacement of the pancreas (38, 41).
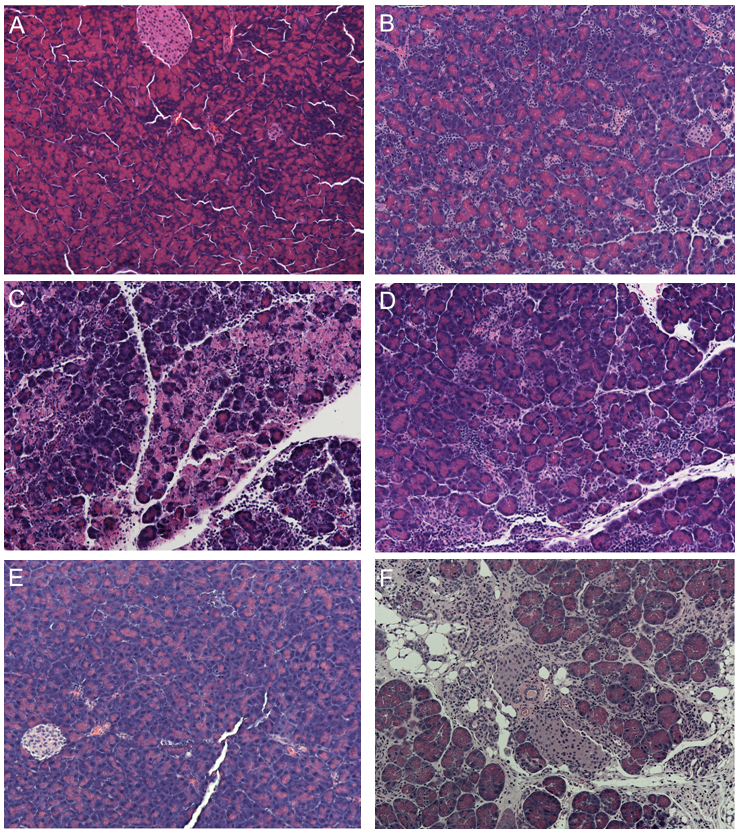
Figure 2. Pancreatic Histology After CVB3/CO Infection of C57BK/6 Mice. Mice were infected with 5 X 105 PFU/ml of CVB3/CO. The animals were euthanized at the indicated times after infection and the pancreata harvested. The pancreata were fixed, and sections stained with hematoxylin & eosin. A) uninfected animal, B) 2 days after infection inflammation is detected, C) 4 days after infection edema, inflammation and acinar cell necrosis is apparent, D) 8 days after infection sign of repair are apparent with the increased presence of acinar cells and acini, E) 12 days after infection much of the acute damage is resolved and many lobes of the pancreas appear normal, F) 14 days after infection accumulation of intrapancreatic fat is apparent.
The accumulation of fat in the pancreata of C57BL/6 mice infected with CVB3/CO may be a useful model for non-alcoholic fatty pancreas disease (NAFPD). Recently, NAFPD has been characterized and associated with diminished pancreatic function and elevated risk of diabetes mellitus (22, 43). A large cross-sectional study found that fatty pancreas was present in 35% of patients presenting for a medical check-up, demonstrating its prevalence (18).
Importantly, CVB3 infection normally spares pancreatic islets. Thus, use of this model allows for the study of both exocrine and endocrine disorders of the pancreas in the presence of increased intrapancreatic fat.
Coxsackieviruses have been shown to establish persistent infection with diminished cytopathology in the hearts of experimentally infected mice and naturally infected humans, as well as in the pancreata of infected mice (5, 15, 35). Thus, it is likely that CVB3 can also persist in the human pancreas. Persistent CVB3 infection of the pancreas may have a role in a number of diseases of the pancreas. For example, elevated antibody titers to coxsackievirus have been reported in patients with chronic pancreatitis (23). Conversely, it is possible that persistent CVB3 infection perpetuates a low level inflammatory response or that the continued expression of coxsackievirus proteins alters important cellular functions. Therefore, it is possible that persistent CVB3 infection may be involved in other pancreatic diseases such as autoimmune pancreatitis and pancreatic cancer (5). Additionally, the accumulation of fat in the pancreata of C57BL/6 mice infected with CVB3/CO long after the resolution of the acute pancreatic infection could possibly be related to persistent CVB3 infection of the pancreas.
While NAFPD has recently emerged as a significant factor in pancreatic dysfunction, alcohol abuse has long been associated with both acute and chronic pancreatitis. Alcohol abuse is considered the etiology in approximately 35% of cases of acute pancreatitis in adults (39). It is accepted that alcohol abuse alone is insufficient to incite acute pancreatitis, and, rather than initiate pancreatitis, alcohol serves to sensitize the pancreas to necro-inflammatory disease (1, 24, 29, 44). Our lab has combined the CVB3-induced model of acute pancreatitis with ethanol administration to mice. We have found that ethanol administration exacerbates CVB3-induced pancreatitis (8, 14, 30). Of particular importance, use of CVB3-induced acute pancreatitis as a model for alcoholic acute pancreatitis is physiologically relevant to humans, combining ethanol administration with a trigger known to cause acute pancreatitis in humans.
In summary, coxsackieviruses can cause pancreatitis in mice. The extent of the tissue damage and the regeneration of the pancreatic tissue is dependent on the genetic background of the mouse and the strain of the virus. CVB3-induced acute pancreatitis is physiologically relevant to human acute pancreatitis because it utilizes human pathogens known to cause pancreatitis. Induction of CVB3-induced acute pancreatitis is simple, requiring only a single IP injection, avoids invasive procedures, is highly reproducible, and has a low incidence of complications leading to mortality. CVB3 causes a more severe form of acute pancreatitis than the commonly used model of cerulean administration and thus, may have greater utility in studies investigating the efficacy of pharmacologic agents to treat or prevent progression of this disease. This infectious mode of initiating acute pancreatitis is physiologically relevant to cases of human acute pancreatitis, utilizing a human pathogen not adapted to animals. Currently, treatment options for acute pancreatitis are limited and mortality rates have not improved appreciably for several decades; this has been attributed, in part, to the lack of animal models that recapitulate the pathophysiology of human disease (31). Thus, the relevance of this model to human pathophysiology makes CVB3-induced acute pancreatitis an appealing preclinical model.
5. References
- Apte MV, Pirola RC and Wilson JS. Mechanisms of alcoholic pancreatitis. J Gastroenterol Hepatol 25(12): 1816-1826, 2010. PMID: 21091991.
- Arnesjo B, Eden T, Ihse I, Nordenfelt E and Ursing B. Enterovirus infections in acute pancreatitis - a possible etiological connection. Scand J Gastroenterol 11(7): 645-649, 1976. PMID: 996429.
- Benifla M and Weizman Z. Acute pancreatitis in childhood: analysis of literature data. J Clin Gastroenterol 37(2): 169-172, 2003. PMID: 12869890.
- Capner P, Lendrum R, Jeffries DJ and Walker G. Viral antibody studies in pancreatic disease. Gut 16(11): 866-870, 1975. PMID: 1193416.
- Chapman NM and Kim KS. Persistent coxsackievirus infection: enterovirus persistence in chronic myocarditis and dilated cardiomyopathy. Curr Top Microbiol Immunol 323: 275-292, 2008. PMID: 18357775.
- Chen Y, Zak Y, Hernandez-Boussard T, Park W and Visser BC. The epidemiology of idiopathic acute pancreatitis, analysis of the nationwide inpatient sample from 1998 to 2007. Pancreas 42(1): 1-5, 2013. PMID: 22750972.
- Chrysos G, Kokkoris S, Protopsaltis J, Korantzopoulos P and Giannoulis G. Coxsackievirus infection associated with acute pancreatitis. JOP 5(5): 384-387, 2004. PMID: 15365208.
- Clemens DL and Jerrells TR. Ethanol consumption potentiates viral pancreatitis and may inhibit pancreas regeneration: preliminary findings. Alcohol 33(3): 183-189, 2004. PMID: 15596086.
- Coplan NL, Atallah V, Mediratta S, Bruno MS and DePasquale NP. Cardiac, pancreatic, and liver abnormalities in a patient with coxsackie-B infection. Am J Med 101(3): 325-326., 1996. PMID.
- De Palma AM, Thibaut HJ, Li S, Van Aelst I, Dillen C, Swinnen M, et al. Inflammatory rather than infectious insults play a role in exocrine tissue damage in a mouse model for coxsackievirus B4-induced pancreatitis. J Pathol 217(5): 633-641, 2009. PMID: 19142976.
- Fechner RE, Smith MG and Middlekamp JN. Coxsackie B virus infection of the newborn. Am J Pathol 42: 493-505, 1963. PMID: 13944652.
- Gauntt CJ, Gomez PT, Duffey PS, Grant JA, Trent DW, Witherspoon SM, et al. Characterization and myocarditic capabilities of coxsackievirus B3 variants in selected mouse strains. J Virol 52(2): 598-605, 1984. PMID: 6092681.
- Gomez RM, Lascano EF and Berria MI. Murine acinar pancreatitis preceding necrotizing myocarditis after Coxsackievirus B3 inoculation. J Med Virol 35(2): 71-75., 1991. PMID.
- Jerrells TR, Chapman N and Clemens DL. Animal model of alcoholic pancreatitis: role of viral infections. Pancreas 27(4): 301-304, 2003. PMID.
- Kim KS, Tracy S, Tapprich W, Bailey J, Lee CK, Kim K, et al. 5'-Terminal deletions occur in coxsackievirus B3 during replication in murine hearts and cardiac myocyte cultures and correlate with encapsidation of negative-strand viral RNA. J Virol 79(11): 7024-7041, 2005. PMID: 15890942.
- Lal SM, Fowler D, Losasso CJ and Berg GG. Coxsackie virus-induced acute pancreatitis in a long-term dialysis patient. Am J Kidney Dis 11(5): 434-436, 1988. PMID: 2835903.
- Lerch MM and Gorelick FS. Models of acute and chronic pancreatitis. Gastroenterology 144(6): 1180-1193, 2013. PMID: 23622127.
- Lesmana CR, Pakasi LS, Inggriani S, Aidawati ML and Lesmana LA. Prevalence of Non-Alcoholic Fatty Pancreas Disease (NAFPD) and its risk factors among adult medical check-up patients in a private hospital: a large cross sectional study. BMC Gastroenterol 15: 174, 2015. PMID: 26652175.
- Mena I, Fischer C, Gebhard JR, Perry CM, Harkins S and Whitton JL. Coxsackievirus infection of the pancreas: evaluation of receptor expression, pathogenesis, and immunopathology. Virology 271(2): 276-288, 2000. PMID: 10860882.
- Murphy AM and Simmul R. Coxsackie B4 Virus Infections in New South Wales during 1962. Med J Aust 2: 443-445, 1964. PMID: 14201721.
- Nakao T, Nitta T, Miura R, Ogata K, Kume T, Nobuta K, et al. Clinical and Epidemiological Studies on an Outbreak of Aseptic Meningitis Caused by Coxsackie B5 and A9 Viruses in Aomori in 1961. Tohoku J Exp Med 83: 94-102, 1964. PMID: 14187867.
- Ou HY, Wang CY, Yang YC, Chen MF and Chang CJ. The association between nonalcoholic fatty pancreas disease and diabetes. PLoS ONE 8(5): e62561, 2013. PMID: 23671610.
- Ozsvar Z, Deak J and Pap A. Possible role of Coxsackie-B virus infection in pancreatitis. Int J Pancreatol 11(2): 105-108, 1992. PMID: 1318913.
- Pandol SJ, Periskic S, Gukovsky I, Zaninovic V, Jung Y, Zong Y, et al. Ethanol diet increases the sensitivity of rats to pancreatitis induced by cholecystokinin octapeptide. Gastroenterology 117(3): 706-716., 1999. PMID.
- Pandol SJ, Saluja AK, Imrie CW and Banks PA. Acute pancreatitis: bench to the bedside. Gastroenterology 132(3): 1127-1151, 2007. PMID: 17383433.
- Parenti DM, Steinberg W and Kang P. Infectious causes of acute pancreatitis. Pancreas 13(4): 356-371, 1996. PMID: 8899796.
- Ramsingh AI, Lee WT, Collins DN and Armstrong LE. Differential recruitment of B and T cells in coxsackievirus B4-induced pancreatitis is influenced by a capsid protein. J Virol 71(11): 8690-8697., 1997. PMID.
- Ramsingh AI, Lee WT, Collins DN and Armstrong LE. T cells contribute to disease severity during coxsackievirus B4 infection. J Virol 73(4): 3080-3086., 1999. PMID.
- Sata N, Koizumi M and Nagai H. Alcoholic pancreatopathy: a proposed new diagnostic category representing the preclinical stage of alcoholic pancreatic injury. J Gastroenterol 42 Suppl 17: 131-134, 2007. PMID: 17238042.
- Schneider KJ, Scheer M, Suhr M and Clemens DL. Ethanol administration impairs pancreatic repair after injury. Pancreas 41(8): 1272-1279, 2012. PMID: 22617711.
- Su KH, Cuthbertson C and Christophi C. Review of experimental animal models of acute pancreatitis. HPB (Oxford) 8(4): 264-286, 2006. PMID: 18333137.
- Suzuki M, Sai JK and Shimizu T. Acute pancreatitis in children and adolescents. World J Gastrointest Pathophysiol 5(4): 416-426, 2014. PMID: 25400985.
- Tracy S, Drescher KM, Chapman NM, Kim KS, Carson SD, Pirruccello S, et al. Toward testing the hypothesis that group B coxsackieviruses (CVB) trigger insulin-dependent diabetes: inoculating nonobese diabetic mice with CVB markedly lowers diabetes incidence. J Virol 76(23): 12097-12111, 2002. PMID: 12414951.
- Tracy S, Hofling K, Pirruccello S, Lane PH, Reyna SM and Gauntt CJ. Group B coxsackievirus myocarditis and pancreatitis: connection between viral virulence phenotypes in mice. J Med Virol 62(1): 70-81, 2000. PMID.
- Tracy S, Smithee S, Alhazmi A and Chapman N. Coxsackievirus can persist in murine pancreas by deletion of 5' terminal genomic sequences. J Med Virol 87(2): 240-247, 2015. PMID: 25111164.
- Vonlaufen A, Wilson JS and Apte MV. Molecular mechanisms of pancreatitis: current opinion. J Gastroenterol Hepatol 23(9): 1339-1348, 2008. PMID: 18853993.
- Vuorinen T, Kallajoki M, Hyypia T and Vainionpaa R. Coxsackievirus B3-induced acute pancreatitis: analysis of histopathological and viral parameters in a mouse model. Br J Exp Pathol 70(4): 395-403, 1989. PMID: 2548562.
- Walters MN, Leak PJ, Joske RA, Stanley NF and Perret DH. Murine Infection with Reovirus. 3. Pathology of Infection with Types 1 and 2. Br J Exp Pathol 46: 200-212, 1965. PMID: 14286949.
- Wang GJ, Gao CF, Wei D, Wang C and Ding SQ. Acute pancreatitis: etiology and common pathogenesis. World J Gastroenterol 15(12): 1427-1430, 2009. PMID: 19322914.
- Whitcomb DC. Clinical practice. Acute pancreatitis. N Engl J Med 354(20): 2142-2150, 2006. PMID: 16707751.
- Witte CL and Schanzer B. Pancreatitis due to mumps. JAMA 203(12): 1068-1069, 1968. PMID: 5694373.
- Wolfgram LJ, Beisel KW, Herskowitz A and Rose NR. Variations in the susceptibility to Coxsackievirus B3-induced myocarditis among different strains of mice. J Immunol 136(5): 1846-1852, 1986. PMID: 3005402.
- Wu WC and Wang CY. Association between non-alcoholic fatty pancreatic disease (NAFPD) and the metabolic syndrome: case-control retrospective study. Cardiovasc Diabetol 12: 77, 2013. PMID: 23688357.
- Yadav D, Papachristou GI and Whitcomb DC. Alcohol-associated pancreatitis. Gastroenterol Clin North Am 36(2): 219-238, 2007. PMID: 17533076.
- Yap IS, Giddings G, Pocock E and Chantler JK. Lack of islet neogenesis plays a key role in beta-cell depletion in mice infected with a diabetogenic variant of coxsackievirus B4. J Gen Virol 84(Pt 11): 3051-3068, 2003. PMID: 14573810.
- Yoon JW, Austin M, Onodera T and Notkins AL. Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N Engl J Med 300(21): 1173-1179, 1979. PMID: 219345.