
Methods Type:
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2010.13
| Attachment | Size |
|---|---|
| 270.54 KB |
1. Introduction
Autophagy refers to any cellular degradative pathway that involves the delivery of cytoplasmic cargo to the lysosome. Three forms of autophagy have been identified so far: chaperone-mediated autophagy, microautophagy, and macroautophagy that differ with respect to their physiological functions and the mode of cargo delivery to the lysosome. Chaperone-mediated autophagy involves the direct translocation of cytosolic proteins across the lysosomal membrane, which requires protein unfolding by chaperone proteins. Microautophagy involves inward invagination of lysosomal membrane, which delivers a small portion of cytoplasm into the lysosomal lumen. Macroautophagy (simply referred to as autophagy hereafter), is the major regulated catabolic mechanism that eukaryotic cells use to degrade long-lived proteins and organelles. This form of autophagy involves the delivery of cytoplasmic cargo sequestered inside double-membrane vesicles to the lysosome. Initial steps include the formation (vesicle nucleation) and expansion (vesicle elongation) of an isolation membrane, which is also called a phagophore. Then, the edges of the phagophore fuse (vesicle completion) to form the autophagosome, a double-membraned vesicle that sequesters the cytoplasmic material. This is followed by fusion of the autophagosome with a lysosome to form an autolysosome where the captured material, together with the inner membrane, is degraded (37, 46).
Autophagy occurs at low basal levels in virtually all cells to perform homeostatic functions such as protein and organelle turnover. It is rapidly upregulated when cells need generation of intracellular nutrients and energy, for example during starvation, growth factor withdrawal or high bioenergetic demand. Autophagy is also upregulated when cells are preparing for structural remodeling such as during developmental transitions or to rid themselves of damaging cytoplasmic components, such as during oxidative stress, infection or protein aggregate accumulation. Nutritional status, hormonal factors and other parameters such as temperature, oxygen concentration and cell density are important in the control of autophagy.
One of the key regulators of autophagy is the target of rapamycin, TOR kinase (TOR in yeast, mTOR in mammalian cells), which is the major inhibitory signal that shuts off autophagy in the presence of growth factors and abundant nutrients. The class I PI3K/Akt signaling molecules link receptor tyrosine kinases to TOR activation and thereby repress autophagy in response to insulin-like and other growth factor signals. Some of the other regulatory molecules that control autophagy include 5’-AMP-activated protein kinase (AMPK), which responds to energy depletion, the eukaryotic initiation factor 2α (eIF2α), which responds to nutrient starvation, double-stranded RNA and endoplasmic reticulum (ER) stress, BH3-only proteins that contain a Bcl-2 homology-3 (BH3) domain which disrupt Bcl-2/Bcl-XL complex; the tumor suppressor protein p53, death-associated protein kinases (DAPk), the ER-membrane-associated protein Ire-1, the stress-activated kinase c-Jun-N-terminal kinase, the inositoltrisphosphate (IP3) receptor (IP3R), GTPases, Erk1/2, ceramide and calcium (3, 24, 30).
In yeast, more than 30 genes downstream from the TOR kinase (known as the ATG genes) encode proteins (many of which are conserved in evolution) that are essential for the execution of autophagy. These include a protein serine/threonine kinase complex that responds to upstream signals such as TOR kinase (Atg1, Atg13, Atg17), a lipid kinase signaling complex that mediates vesicle nucleation (Atg6, Atg14, Vps34 and Vps15), two ubiquitin-like conjugation pathways that mediate vesicle expansion (the Atg8 and Atg12 systems), a recycling pathway that mediates the disassembly of Atg proteins from mature autophagosomes (Atg2, Atg9, Atg18) and vacuolar permeases that permit the efflux of amino acids from the degradative compartment (Atg22). In mammals, proteins that act more generally in lysosomal function are required for proper fusion with autophagosomes, such as the lysosomal transmembrane proteins, LAMP-2 and CLN3, and for the degradation of autophagosomal contents, such as the lysosomal cysteine proteases, cathepsins B, D and L (2, 49).
The identification of signals that regulate autophagy and of genes that execute autophagy has facilitated detection and manipulation of the autophagy pathway. Phosphatidylethanolamine (PE) conjugation of yeast Atg8 or mammalian LC3 during autophagy results in a nonsoluble form of Atg8 (Atg8-PE) or LC3 (LC3-II) that stably associates with the autophagosomal membrane. Consequently, autophagy can be detected biochemically, by assessing the generation of Atg8-PE or LC3-II, or microscopically, by observing the localization pattern of fluorescently tagged Atg8 or LC3 (see below). These approaches must be coupled with additional measures to discriminate between two physiologically distinct scenarios, increased autophagic flux without impairment in autophagic turnover versus impaired clearance of autophagosomes, which results in a functional defect in autophagic catabolism and can lead to their accumulation (16, 22, 39).
Autophagy can be pharmacologically induced by inhibiting negative regulators, such as TOR with rapamycin, the antiapoptotic proteins Bcl-2 and Bcl-XL which bind to the mammalian ortholog of yeast Atg6, Beclin 1, with ABT-737, IP3R with xestospongin B, which raises the level of the IP3R antagonist, or lithium, a molecule that decreases IP3 activity. Autophagy can be pharmacologically inhibited by targeting the class III PI3K involved in autophagosome formation with 3-methyladenine (42), by targeting the fusion of autophagosomes with lysosomes using vimblastine or by the inhibition of lysosomal hydrolases using inhibitors of the lysosomal proton pump such as bafilomycin A1 or lysosomotropic alkalines such as chloroquine and 3-hydroxychloroquine (21, 43, 47). It should be noted that all of these pharmacological agents lack specificity for the autophagy pathway. Therefore, although some of these agents such as rapamycin, lithium and chloroquine are clinically available and may be helpful for treating diseases associated with autophagy deregulation, genetic approaches to inhibiting autophagy, for example the knockout of ATG genes by homologous recombination or their knockdown by small interfering RNA (siRNA), have yielded more conclusive information about the biologic roles of autophagy in health and disease.
Under physiological conditions, autophagy has a number of vital roles such as maintaining the amino acid pool during starvation, preimplantation development, prevention of neurodegeneration, antiaging, tumor suppression, clearance of intracellular microbes and regulation of innate and adaptive immunity. The best inducer of autophagy is nutrient starvation, both in cultured cells and in intact organisms, ranging from yeast to mammals. Besides starvation, autophagy can also be activated by other physiological stress stimuli (e.g., hypoxia, energy depletion, endoplasmic reticulum stress, high temperature and high-density conditions), hormonal stimulation, pharmacological agents (e.g., rapamycin and other compounds listed below), innate immune signals and in diseases such as bacterial, viral, and parasitic infections, protein aggregopathies, heart disease and acute pancreatitis. Conversely, autophagy suppression is also often associated with certain diseases, including a subset of cancers, neurodegenerative disorders, infectious diseases and inflammatory bowel disorders, and a decline in autophagy function is a common feature of aging.
The molecular cascade that regulates and executes autophagy has been the subject of recent, comprehensive reviews (11, 18, 20, 23, 25-27, 31-36, 41, 44, 45, 48)
2. Autophagy in the pancreas
Helin et al. (12) provided the first morphologic evidence of autophagy in human pancreatitis. Actually, it is one of the earliest descriptions of autophagy in a pathological processes. Autophagy was first identified in mammalian liver upon glucagon treatment approximately 50 years ago, but its molecular understanding started only in the past decade, largely based on the discovery of autophagy-related (ATG) genes. Among the Atg proteins, one subset is essential for autophagosome formation in mammalian tissue, and is referred to as the “core” molecular machinery (14). These core Atg proteins are composed of four subgroups: first, the Atg1/unc-51-like kinase (ULK) complex; second, two ubiquitin-like protein (Atg12 and Atg8/LC3) conjugation systems; third, the class III phosphatidylinositol 3-kinase (PtdIns3K)/Vps34 complex I; and fourth, two transmembrane proteins, Atg9/mAtg9 and VMP1. Mammalian Atg9 (mAtg9) and vacuole membrane protein 1 (VMP1) (40) are two transmembrane proteins so far identified that are required for mammalian autophagy. In contrast to mAtg9, VMP1, which was described in pancreas during experimental acute pancreatitis, has no known homolog in yeast (4). Actually, autophagosomes labeled by the VMP1-LC3 colocalization technique were reported in pancreatic tissue from rats with acute pancreatitis (40). The current knowledge has led to the proposal that autophagy is involved in the pathogenia of acute pancreatitis. However, several mechanisms were proposed to explain its role. For instance, Hashimoto et al. (10) showed that acute pancreatitis was not observed in conditional knockout mice lacking the autophagy-related 5 gene in acinar cells suggesting that an excess in autophagy induces pancreatitis. On the other hand, using a rat model of acute pancreatitis induced by chronic alcohol intake and acute endotoxemia, Fotunato et al (6) reported the accumulation of autophagosomes without an increase in autolysosomes, coupled to the depletion of LAMP-2, a lysosomal protein required for the proper fusion of autophagosomes with lysosomes. Finally, Mareninova et al. proposed that autophagy is retarded in pancreatitis due to deficient lysosomal degradation caused by impaired cathepsin processing (28). Autophagy is also involved in Diabtes mellitus. Moreover, an active role was proposed for autophagy in the pathophysiology of diabetes mellitus since an increase in the number of dead β-cells in diabetic islets compared to non-diabetic cases was recently observed in pancreatic samples from individuals with type 2 diabetes (29). Autophagy and VMP1 expression were found in pancreatic beta cells during experimental diabetes (9). Finally, autophagy-related phenotypic changes have been described in pancreatic tumor cells. Pancreatic tumor cells activate autophagy in response to microenvironment changes associated with cancer, such as hypoxia and low nutrient supply. Autophagic features are reported in peripheral areas of surgically resected pancreatic cancers (7). Moreover, up-regulation of autophagy favors survival of pancreatic cancer cells in tumor-associated inflammation. Over-expression of RAGE is associated with enhanced autophagy, decreased apoptosis and increased pancreatic cancer cell viability (15). On the other hand, repression of autophagy-mediated cell death enhances tumor cell survival and explains in part pancreatic cancer resistance to treatments (1). Finally, autophagy is a mechanism of tumor cell death in response to chemotherapeutic drugs and several agents such as Cardiac Glycosides (5), gemcitabine (38) and Sorafenib (50) exert their effect at least in part through promotion of autophagy. These observations point at autophagy induction as a promising tool for pancreatic cancer therapy.
3. Methods
During the last years there has been a burst of research on autophagy. A common misconception that must be taken into account is that an increased number of autophagosomes in cells corresponds to increased cellular autophagic activity. Given that the autophagosome is an intermediate structure in a dynamic pathway, the number of autophagosomes observed at any specific time point is a function of the balance between the rate of their generation and the rate of their conversion into autolysosomes. Thus, autophagosome accumulation may represent either autophagy induction or, alternatively, suppression of steps in the autophagy pathway downstream from autophagosome formation. If any step upstream of autophagosome formation is blocked, the number of all autophagic structures is decreased. In contrast, the blockade of any step downstream of autophagosome formation increases the number of autophagosomes while decreasing the number of autolysosomes. Therefore, the mere determination of autophagosome number is insufficient for an overall estimation of autophagic activity. Rather, different methods often need to be used in concert to distinguish between basal levels of autophagy, induction of autophagy and suppression of upstream or downstream steps of autophagy. The term “autophagic flux” is used to denote the dynamic process of autophagosome synthesis, delivery of autophagic substrates to the lysosome and degradation of autophagic substrates inside the lysosome . Itis a more reliable indicator of autophagic activity than counting autophagosomes. In the following sections, we will discuss different methods for monitoring the number of autophagosomes and for monitoring the “autophagic flux” in pancreatic cells as well as in other cell types.
1. Quantifying the number of autophagosomes
Three methods are currently used to quantify the number of autophagosomes: a) electron microscopy, b) subcellular localization of LC3, and c) detection of processed LC3.
1a. Electron microscopy
The most traditional method of counting is electron microscopy. At the ultrastructural level, an autophagosome is defined as a double-membraned structure containing undigested cytoplasmic contents, which has not fused with a lysosome. Autophagosomes often enclose intracellular organelles such as mitochondria and fragments of the ER. As this definition is straightforward, it is usually easy to identify autophagosomes, or at least those organelles that envelop cellular contents. In contrast to an autophagosome containing cellular cargo (which is generally easy to identify), the distinction of autolysosomes from other cellular membranous compartments is frequently more complicated. The autolysosome is a hybrid organelle generated by the fusion of an autophagosome and a lysosome, which has a single limiting membrane and contains cytoplasmic materials at various stages of degradation. At early stages, the inside materials can be recognized as having originated from cytoplasm. However, if degradation proceeds too far, it is not easy to determine whether the inside materials are of intracellular origin.
1b. Subcellular localization of LC3
The assessment of autophagosome number by electron microscopy is expensive, time consuming and requires considerable specialized expertise. It is becoming increasingly replaced by light microscopic analysis and biochemical methods that are more widely accessible to researchers in different fields. The mammalian autophagy protein, LC3, is a marker of autophagosomes. Among the four LC3 isoforms, LC3B is most widely used. After its synthesis, nascent LC3 is processed at its C terminus by Atg4 and becomes LC3-I, which has a glycine residue at the C-terminal end. LC3-I is subsequently conjugated with phosphatidylethanolamine to become LC3-II (LC3-PE) by a ubiquitination-like enzymatic reaction. In contrast to the cytoplasmic localization of LC3-I, LC3-II associates with both the outer and inner membranes of the autophagosome. After fusion with the lysosome, LC3 on the outer membrane is cleaved off by Atg4 and LC3 on the inner membrane is degraded by lysosomal enzymes, resulting in very low LC3 content in the autolysosome. Thus, endogenous LC3 or GFP-LC3 (GFP-LC3 plasmid can be obtained at http://www.addgene.org) is visualized by fluorescence microscopy either as a diffuse cytoplasmic pool or as punctate structures that primarily represent autophagosomes (see Figure 1). Although the number of punctate LC3 or GFP-LC3 structures per cell is usually an accurate measure of autophagosome number, this assay has some potential experimental pitfalls. First, there is potential for subjectivity, and a uniform approach needs to be established and applied by the investigator, both with respect to the method of quantitation and the criteria for defining a “punct”. Although the number of puncta is markedly increased after autophagy induction, small numbers of puncta are also observed even under normal conditions. Therefore, “percentage of cells with GFP-LC3 puncta” is not an appropriate indicator; if so, results should then be expressed as the “percentage of cells with more than a certain number of puncta.” In general, it is preferable to quantify the “average number of GFP-LC3 puncta per cell” in all cells in the population under evaluation. A second potential pitfall with the detection of LC3 or GFP-LC3 punctate structures as a method to monitor autophagosome numbers is the observation that GFP-LC3, and probably even endogenous LC3, can be easily aggregated if overexpressed or coexpressed with other aggregate-prone proteins. GFP-LC3 aggregates are often indistinguishable by fluorescence microscopy from true autophagosomes. However, certain precautions can be exercised to reduce the possibility of GFP-LC3 aggregation. The use of stable GFP-LC3 transformants is highly recommended, so that one can select clones that express appropriate levels of GFP-LC3 without artificial aggregation. When the GFP-LC3 construct is used in transient transfection experiments, caution should be exercised to avoid high levels of expression that result in artificial aggregation. The GFP-LC3 labeling method has been successfully applied to in vivo mammalian autophagy research by generating transgenic GFP-LC3 transgenic mice (which can be obtained at http://www2.brc.riken.jp/lab/animal/detail.cgi?reg_no=00806), Drosophila, nematodes, plants and zebrafish. In GFP-LC3 transgenic mice, GFP-LC3 is ubiquitously expressed under the control of the CAG promoter, and the accumulation of GFP punctae (which represent autophagosomes) is observed in pancreas. In addition to the systemic GFP-LC3 transgenic mice, tissue-specific transgenic mice expressing GFP-LC3 and mCherry-LC3 have also been generated. These systemic and tissue-specific models have been effectively used to show reductions in autophagosome numbers in mice deficient in autophagy genes. They have also been successfully used to show increases in autophagosome numbers under disease and stress conditions. Thus, the monitoring of GFP-LC3 punctae in GFP-LC3 transgenic mice is a powerful method to assess whether different physiological and pathophysiological stimuli regulate autophagosome numbers in vivo.
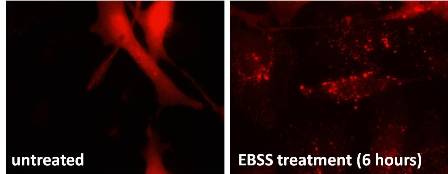
Figure 1. HeLa cells were transfected with a GFP-LC3 plasmid and 24 later cultivated in Earles Balanced Salt Solution (EBSS), an autophagy-inducing medium due to the lack of glucose and amino acids, or DMEM, a complete media, for 6 additional hours. Dots correspond to autophagosomes.
1c. Detection of the processed LC3
In addition to its utility in fluorescence microscopy assays, LC3 is also useful in biochemical assays to assess autophagosome numbers. The conversion from endogenous LC3-I to LC3-II can be detected by immunoblotting with antibodies against LC3 (LC3 antibodies from MBL International (ref #PM036), from Nanotools (ref #0261-100/LC3-5H3) or from Santa Cruz (ref # 16756) give satisfactory results when diluted at 1:500 and in TBS/1% BSA buffer). Although the actual molecular weight of LC3-II (the PE conjugated form) is larger than that of LC3-I, LC3-II migrates faster than LC3-I in SDS-PAGE because of extreme hydrophobicity of LC3-II. The amount of LC3-II usually correlates well with the number of autophagosomes (see Figure 2). However, not all LC3-II is present on autophagic membranes, and, importantly, some population of LC3-II seems to be ectopically generated in an autophagy-independent manner. For example, a significant amount of LC3-II is detectable in FIP200- and Atg14-deficient mouse embryonic fibroblasts, in Beclin 1-deficient embryonic stem cells and in cells with RNA interference (RNAi)-mediated suppression of Beclin 1, Atg13, Atg14, and Vps34, even though autophagosome formation is completely or profoundly inhibited. Therefore, in settings where certain components of the autophagic machinery are genetically or pharmacologically inactivated, it is still possible that autophagy is suppressed even if LC3-II is detected. In such cases, other approaches, including “autophagic flux” assays are required to assess autophagic activity.
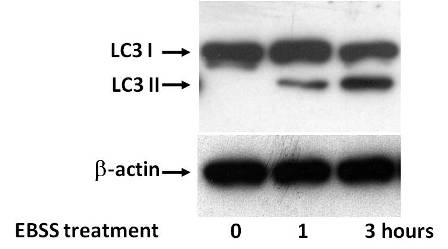
Figure 2. Panc1 pancreatic cancer cells were cultivated in EBSS autophagy-inductor media for 1 and 3 hours and cleavage of endogenous LC3 measured by western blot. β-actin was used as control.
2. Measuring “autophagic flux”
As discussed above, the accumulation of autophagosomes is not always indicative of autophagy induction and may represent either the increased generation of autophagosomes and/or a block in autophagosomal maturation and the completion of the autophagy pathway. In most experimental settings, it is necessary to distinguish whether autophagosome accumulation is due to autophagy induction or rather a block in downstream steps, by performing “autophagic flux” assays that distinguish between these two possibilities. Several assays are presently available to monitor “autophagic flux” by measuring: a) LC3 turnover, b) levels of autophagic substrates, c) mRFP-GFP-LC3 color change, d) free GFP generated from GFP-LC3, and e) lysosome-dependent long-lived protein degradation.
2a. LC3 turnover
One of the principal methods in current use to measure “autophagic flux” is the monitoring of LC3 turnover, which is based on the observation that LC3-II is degraded in autolysosomes. In fact, in cells treated with lysosomotropic reagents such as ammonium chloride, chloroquine or bafilomycin A1, which inhibit acidification inside the lysosome or inhibit autophagosome-lysosome fusion, or with inhibitors of lysosomal proteases such as E64d and pepstatin A, the degradation of LC3-II is blocked, resulting in the accumulation of LC3-II. Accordingly, the differences in the amount of LC3-II between samples in the presence and absence of lysosomal inhibitors represent the amount of LC3 that is delivered to lysosomes for degradation.
2b. Levels of autophagic substrates
As LC3 is degraded by autophagy, the disappearance of total LC3 is paradoxically a good indicator of autophagic flux. Even the amount of LC3-II, which increases transiently upon induction of autophagy, is decreased after longer periods of autophagy activation. Besides LC3, levels of other autophagy substrates can be used to monitor “autophagic flux”. Classically, autophagy was considered to be a random degradation system, but recent studies have revealed that several specific substrates are preferentially degraded by autophagy, of which the best studied example is p62, which is also known as SQSTM1 or sequestome 1. p62 is selectively incorporated into autophagosomes through direct binding to LC3 and is efficiently degraded by autophagy. Thus, the total cellular expression levels of p62 inversely correlate with autophagic activity (see Figure 3). In numerous studies, the measurement of cellular p62 appears to correlate well with other parameters of “autophagic flux”, and overall, this assay seems quite promising (p62 antibodies from Abgent (ref # AP2183b) or from Cell Signaling (ref # 5114S) give satisfactory results when diluted at 1:500 and in TBS/1% BSA buffer). However, p62 as well as LC3, can be transcriptionally regulated during autophagy, which may confound the interpretation of p62 and LC3 levels as indicators of “autophagic flux”.
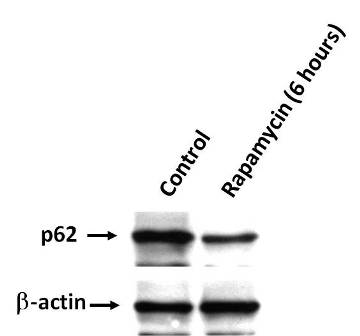
Figure 3. Panc1 pancreatic cancer cells were treated with Rapamycin by a 6-hours period and p62 amount was measured by western blot. β-actin was used as control.
2c. mRFP-GFP-LC3 color change
Another useful assay to measure “autophagic flux” is based on the concept of lysosomal quenching of GFP in GFP-labeled autophagic substrates such as LC3. GFP is a stably folded protein and relatively resistant to lysosomal proteases. However, the low pH inside the lysosome quenches the fluorescent signal of GFP, which makes it difficult to trace the delivery of GFP-LC3 to lysosomes. Indeed, most GFP-LC3 punctate signals do not colocalize with lysosomes. In contrast, RFP or mCherry exhibits more stable fluorescence in acidic compartments and mRFP-LC3 can readily be detected in autolysosomes. By exploiting the difference in the nature of these two fluorescent proteins (i.e., lysosomal quenching of GFP fluorescence versus lysosomal stability of RFP fluorescence), “autophagic flux” can be morphologically traced with an mRFP-GFP-LC3 tandem construct (mRFP-GFP-LC3 construct can be obtained at http://www.addgene.org). With this novel construct, autophagosomes and autolysosomes are labeled with yellow (i.e., mRFP and GFP) and red (i.e., mRFP only) signals, respectively. If “autophagic flux” is increased, both yellow and red punctae are increased, however, if autophagosome maturation into autolysosomes is blocked, only yellow punctae are increased without a concomitant increase in red punctae (see Figure 4). Although this assay can be used as an indicator of “autophagic flux”, it does not provide precisely the same information as other flux assays that directly measure endpoints of lysosomal degradation (17).
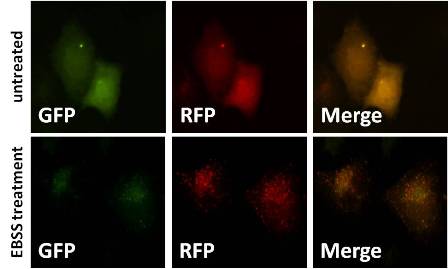
Figure 4. Panc1 pancreatic cancer cells were transfected with mRFP-GFP-LC3 construct and 24 later cultivated in the EBSS autophagy-inductor or DMEM media for 6 additional hours. GFP is quenched in the lysosomal compartment whereas mRFP remains stable. Autophagosomes and autolysosomes are labeled with yellow (i.e., mRFP and GFP) and red (i.e., mRFP only) signals respectively.
2d. Free GFP generated from GFP-LC3
Although GFP fluorescence is quenched by the acidic pH environment inside the lysosome, GFP is still detectable by immunoblotting and is more stable than the GFP-LC3 fusion protein (which is partially degraded upon reaching the lysosome, resulting in the appearance of a free GFP fragment). Therefore, another assay to measure “autophagic flux” is the detection of the free GFP fragment that is generated by degradation of GFP-LC3 in the autolysosome by immunoblotting with an anti-GFP antibody (anti-GFP antibodies from Abcam or from Invitrogen give satisfactory results) (8,13).
2e. Lysosome-dependent long-lived protein degradation
One of the most traditional methods to evaluate “autophagic flux” is the measurement of bulk degradation of long-lived proteins. In this assay, cells are cultured with isotope-labeled amino acids for a long duration (several hours to several days) to label long-lived proteins, followed by a short incubation period without isotope-labeled amino acids to wash out radiolabeled short-lived proteins, which are primarily degraded by the proteasome. After treatment with an autophagy-inducing stimulus, the cellular release of degraded proteins (measured as tricholoracetic acid-soluble radioactivity in the culture supernatant) is quantified. This may be the most quantitative assay, because it provides a precise numerical readout that reflects the fate of all long-lived cellular proteins and avoids the pitfalls associated with measuring a single autophagic substrate. To ensure that one is truly measuring the contribution of “autophagic degradation” (versus other potential pathways that may contribute to long-lived protein degradation), it is standard practice to compare degradation rates between samples cultured in the presence or absence of an autophagy inhibitor such as 3-methyladenine) (19,28,37,42).
4. Conclusions
There is no perfect method to measure autophagosome number or to measure “autophagic flux”. Accordingly, it should be emphasized that there is no single “gold standard” method to monitor or to modulate autophagic activity. Rather, one should consider the use of several concurrent methods (with nonoverlapping limitations) to accurately assess the status and functions of autophagic activity in any given biological setting.
5. References
- Akar U, Ozpolat B, Mehta K, Fok J, Kondo Y, and Lopez-Berestein G. Tissue transglutaminase inhibits autophagy in pancreatic cancer cells. Mol Cancer Res 5: 241-249, 2007. PMID: 17374730
- Bursch W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ 8: 569-581, 2001. PMID: 11536007
- Criollo A, Maiuri MC, Tasdemir E, Vitale I, Fiebig AA, Andrews D, Molgo J, Diaz J, Lavandero S, Harper F, Pierron G, di Stefano D, Rizzuto R, Szabadkai G, and Kroemer G. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ 14: 1029-1039, 2007. PMID: 17256008
- Dusetti NJ, Jiang Y, Vaccaro MI, Tomasini R, Azizi Samir A, Calvo EL, Ropolo A, Fiedler F, Mallo GV, Dagorn JC, and Iovanna JL. Cloning and expression of the rat vacuole membrane protein 1 (VMP1), a new gene activated in pancreas with acute pancreatitis, which promotes vacuole formation. Biochem Biophys Res Commun 290: 641-649, 2002. PMID: 11785947
- Ewald B, Sampath D, and Plunkett W. Nucleoside analogs: molecular mechanisms signaling cell death. Oncogene 27: 6522-6537, 2008. PMID: 18955977
- Fortunato F, Burgers H, Bergmann F, Rieger P, Buchler MW, Kroemer G, and Werner J. Impaired autolysosome formation correlates with Lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology 137: 350-360, 360 e351-355, 2009. PMID: 19362087
- Fujii S, Mitsunaga S, Yamazaki M, Hasebe T, Ishii G, Kojima M, Kinoshita T, Ueno T, Esumi H, and Ochiai A. Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci 99: 1813-1819, 2008. PMID: 18616529
- Gao W, Ding WX, Stolz DB, Yin XM. Induction of macroautophagy by exogenously introduced calcium. Autophagy. 2008 Aug 16;4(6):754-61. PMID: 18560273
- Grasso D, Sacchetti ML, Bruno L, Lo Re A, Iovanna JL, Gonzalez CD, and Vaccaro MI. Autophagy and VMP1 expression are early cellular events in experimental diabetes. Pancreatology 9: 81-88, 2009. PMID: 19077458
- Hashimoto D, Ohmuraya M, Hirota M, Yamamoto A, Suyama K, Ida S, Okumura Y, Takahashi E, Kido H, Araki K, Baba H, Mizushima N, and Yamamura K. Involvement of autophagy in trypsinogen activation within the pancreatic acinar cells. J Cell Biol 181: 1065-1072, 2008. PMID: 18591426
- He C and Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43: 67-93, 2009. PMID: 19653858
- Helin H, Mero M, Markkula H, and Helin M. Pancreatic acinar ultrastructure in human acute pancreatitis. Virchows Arch A Pathol Anat Histol 387: 259-270, 1980. PMID: 7456314
- Hosokawa N, Hara Y, Mizushima N. Generation of cell lines with tetracycline-regulated autophagy and a role for autophagy in controlling cell size. FEBS Lett. 2006 May 15;580(11):2623-9. PMID: 16647067
- Itakura E and Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 6, 2010. PMID: 20639694
- Kang R, Tang D, Schapiro NE, Livesey KM, Farkas A, Loughran P, Bierhaus A, Lotze MT, and Zeh HJ. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ 17: 666-676, 2010. PMID: 19834494
- Kim J, Huang WP, Stromhaug PE, and Klionsky DJ. Convergence of multiple autophagy and cytoplasm to vacuole targeting components to a perivacuolar membrane compartment prior to de novo vesicle formation. J Biol Chem 277: 763-773, 2002. PMID: 11675395
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007 Sep-Oct;3(5):452-60. PMID: 17534139
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 8: 931-937, 2007. PMID: 17712358
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, Yamamoto M, Yue Z, Uchiyama Y, Kominami E, Tanaka K. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007 Dec 14;131(6):1149-63. PMID: 18083104
- Korolchuk VI, Menzies FM, and Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 584: 1393-1398, 2010. PMID: 20040365
- Kovacs AL, Laszlo L, and Kovacs J. Effect of amino acids and cycloheximide on changes caused by vinblastine, leupeptin and methylamine in the autophagic/lysosomal system of mouse hepatocytes in vivo. Exp Cell Res 157: 83-94, 1985. PMID: 3972014
- Kuma A, Matsui M, and Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy 3: 323-328, 2007. PMID: 17387262
- Levine B, Sinha S, and Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy 4: 600-606, 2008. PMID: 18497563
- Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, and Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120: 237-248, 2005. PMID: 15680329
- Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, and Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol 22: 181-185, 2010. PMID: 20044243
- Maiuri MC, Tasdemir E, Criollo A, Morselli E, Vicencio JM, Carnuccio R, and Kroemer G. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ 16: 87-93, 2009. PMID: 18806760
- Maiuri MC, Zalckvar E, Kimchi A, and Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 8: 741-752, 2007. PMID: 17717517
- Mareninova OA, Hermann K, French SW, O'Konski MS, Pandol SJ, Webster P, Erickson AH, Katunuma N, Gorelick FS, Gukovsky I, and Gukovskaya AS. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J Clin Invest 119: 3340-3355, 2009. PMID: 19805911
- Masini M, Bugliani M, Lupi R, del Guerra S, Boggi U, Filipponi F, Marselli L, Masiello P, and Marchetti P. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 52: 1083-1086, 2009. PMID: 19367387
- Meijer AJ and Codogno P. Signalling and autophagy regulation in health, aging and disease. Mol Aspects Med 27: 411-425, 2006. PMID: 16973212
- Mizushima N. Physiological functions of autophagy. Curr Top Microbiol Immunol 335: 71-84, 2009. PMID: 19802560
- Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 22: 132-139, 2010. PMID: 20056399
- Mizushima N and Klionsky DJ. Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr 27: 19-40, 2007. PMID: 17311494
- Mizushima N, Levine B, Cuervo AM, and Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 451: 1069-1075, 2008. PMID: 18305538
- Moreau K, Luo S, and Rubinsztein DC. Cytoprotective roles for autophagy. Curr Opin Cell Biol 22: 206-211, 2010. PMID: 20045304
- Morselli E, Galluzzi L, Kepp O, Vicencio JM, Criollo A, Maiuri MC, and Kroemer G. Anti- and pro-tumor functions of autophagy. Biochim Biophys Acta 1793: 1524-1532, 2009. PMID: 19371598
- Mortimore GE, Hutson NJ, and Surmacz CA. Quantitative correlation between proteolysis and macro- and microautophagy in mouse hepatocytes during starvation and refeeding. Proc Natl Acad Sci U S A 80: 2179-2183, 1983. PMID: 6340116
- Pardo R, Lo Re A, Archange C, Ropolo A, Papademetrio DL, Gonzalez CD, Alvarez EM, Iovanna JL, and Vaccaro MI. Gemcitabine induces the VMP1-mediated autophagy pathway to promote apoptotic death in human pancreatic cancer cells. Pancreatology 10: 19-26, 2010. PMID: 20299819
- Perry CN, Kyoi S, Hariharan N, Takagi H, Sadoshima J, and Gottlieb RA. Novel methods for measuring cardiac autophagy in vivo. Methods Enzymol 453: 325-342, 2009. PMID: 19216914
- Ropolo A, Grasso D, Pardo R, Sacchetti ML, Archange C, Lo Re A, Seux M, Nowak J, Gonzalez CD, Iovanna JL, and Vaccaro MI. The pancreatitis-induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J Biol Chem 282: 37124-37133, 2007. PMID: 17940279
- Rubinsztein DC, Gestwicki JE, Murphy LO, and Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov 6: 304-312, 2007. PMID: 17396135
- Seglen PO and Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A 79: 1889-1892, 1982. PMID: 6952238
- Trout JJ, Stauber WT, and Schottelius BA. Increased autophagy in chloroquine-treated tonic and phasic muscles: an alternative view. Tissue Cell 13: 393-401, 1981. PMID: 7314075
- Virgin HW and Levine B. Autophagy genes in immunity. Nat Immunol 10: 461-470, 2009. PMID: 19381141
- Wang RC and Levine B. Autophagy in cellular growth control. FEBS Lett 584: 1417-1426, 2010. PMID: 20096689
- Xie Z, Nair U, and Klionsky DJ. Dissecting autophagosome formation: the missing pieces. Autophagy 4: 920-922, 2008. PMID: 18719358
- Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, and Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 23: 33-42, 1998. PMID: 9639028
- Yang Z and Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 22: 124-131, 2010. PMID: 20034776
- Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, Hailey DW, Oorschot V, Klumperman J, Baehrecke EH, and Lenardo MJ. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465: 942-946, 2010. PMID: 20526321
- Zhang G, Park MA, Mitchell C, Hamed H, Rahmani M, Martin AP, Curiel DT, Yacoub A, Graf M, Lee R, Roberts JD, Fisher PB, Grant S, and Dent P. Vorinostat and sorafenib synergistically kill tumor cells via FLIP suppression and CD95 activation. Clin Cancer Res 14: 5385-5399, 2008. PMID: 18765530