
Methods Type:
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2021.11
| Attachment | Size |
|---|---|
| 1.78 MB |
The 1996 discovery that mutations in human cationic trypsinogen, encoded by the serine protease 1 (PRSS1) gene, cause hereditary pancreatitis permanently established the central role of trypsin in pancreatitis pathogenesis [1]. Subsequent studies demonstrated that loss-of-function variants in genes that protect against pancreatitis also contribute to genetic susceptibility. Thus, mutations in the serine protease inhibitor Kazal type 1 (SPINK1) gene reduce defensive trypsin inhibition [2] while mutations in the chymotrypsin C (CTRC) gene impair protective trypsinogen degradation [3]. Together, the pathogenic variants in the PRSS1, SPINK1 and CTRC genes and their interactions form the so-called trypsin-dependent pathological pathway of genetic risk in chronic pancreatitis (Figure 1). Details of this mechanistic framework have been reviewed recently [4]. The key concept of this disease model is that excessive intrapancreatic autoactivation of trypsinogen to trypsin drives onset of acute pancreatitis, progression to recurrent acute pancreatitis and eventually to chronic pancreatitis. This progressive sequence of the three disease stages is a hallmark of genetically determined pancreatitis, although phenotypic variability is common.
Despite strong genetic and biochemical evidence in support of the trypsin-dependent pathway in humans, mouse models that recapitulate the mechanistic and phenotypic aspects of the human disease have been lacking until recently. In our laboratory, we initiated a systematic mouse- modeling program in 2013, focusing on the three fundamental elements of pancreatitis risk: (i) increased autoactivation of trypsinogen, (ii) loss of chymotrypsin-mediated protection and (iii) loss of trypsin inhibition. Here, we will review the models characterized and published to date.
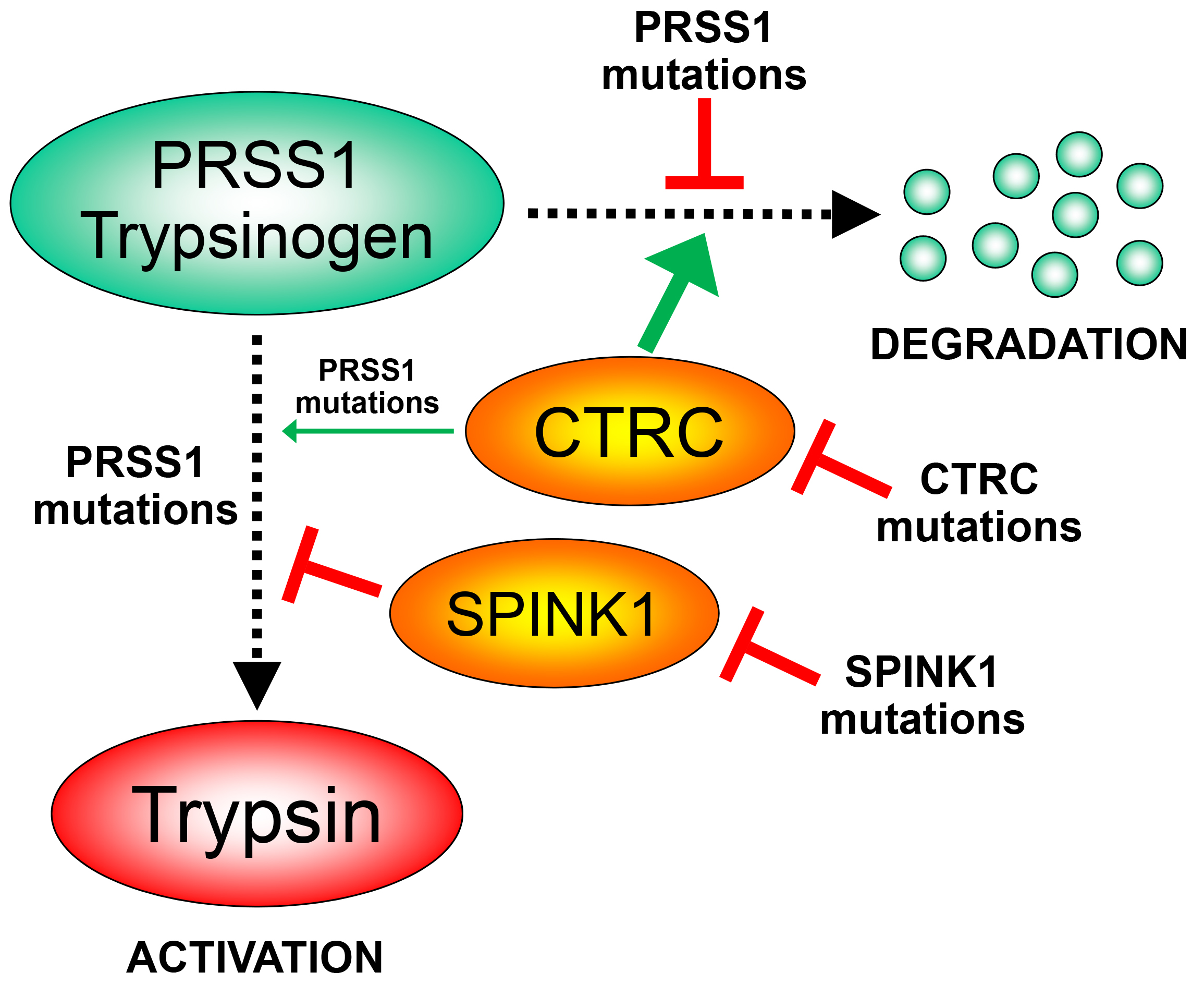
Figure 1. The trypsin-dependent pathway of genetic risk in human chronic pancreatitis. PRSS1 mutations block CTRC-mediated trypsinogen degradation or stimulate trypsinogen autoactivation, either directly or through a CTRC-dependent mechanism. PRSS1, human cationic trypsinogen; CTRC, chymotrypsin C; SPINK1, serine protease inhibitor Kazal type 1.
I. Mouse Models with Increased Trypsinogen Autoactivation
The activation peptide controls trypsinogen activation and mutations in this region often have a marked impact on trypsinogen autoactivation [5]. Although rare, activation-peptide mutations that directly stimulate trypsinogen autoactivation, such as p.D19A, p.D21A, p.D22G, and p.K23R, have been found in human hereditary pancreatitis [6-9]. We developed mouse lines that carry mutations in the activation peptide of the native mouse cationic trypsinogen (isoform T7). We chose this isoform because it is abundantly expressed and has the strongest propensity for autoactivation among mouse trypsinogen isoforms [10]. By targeting the native T7 locus in mice, we ensured that expression levels remained physiological, which is often a problematic aspect of transgenic models. Furthermore, to model the human situation accurately, we generated constitutively expressed mutant alleles. Figure 2 shows the primary structure of the mouse T7 trypsinogen activation peptide with the position of the introduced mutations. For comparison, the activation peptide of human PRSS1 is also shown.
Before introduction into mice, each mutation was thoroughly characterized in the context of purified, recombinant human (PRSS1) and mouse (T7) cationic trypsinogen with respect to autoactivation and activation by cathepsin B [5, 8, 11-14].
The two published models (T7D23A and T7K24R) described below were generated using homologous recombination in mouse embryonic stem (ES) cells. The recombined allele contained a neomycin cassette in intron 1 of the T7 trypsinogen gene, which was removed by breeding the mice with a so-called Cre-deleter strain. This process still leaves a short “scar sequence” in intron 1, which might affect expression. Therefore, we developed a polyclonal anti-peptide antibody specific for T7 trypsinogen and verified that in both strains the expression of mutant T7 trypsinogen was comparable to the wild-type allele. Further details on model generation can be found in the published reports [13, 14]. All our strains are on the C57BL/6N genetic background and as experimental controls we used C57BL/6N mice obtained from Charles River Laboratories or bred in-house from the same stock. The T7D23A strain was maintained in the heterozygous state and freshly purchased C57BL/6N mice were used for breeding. The T7K24R mice were bred to homozygosity and were maintained in this state.
A. The T7D23A mouse model
Properties of these mice have been published in 2018 [13]. The mice carry the p.D23A mutation in the activation peptide of T7 trypsinogen (Figure 2). This mutation is analogous to the human p.D22G mutation found in hereditary pancreatitis [7]. This was one of the first models we designed and to strengthen the phenotype we chose to use an Ala replacement instead of the Gly observed in humans.

Figure 2. Primary structure of the trypsinogen activation peptide in mouse cationic trypsinogen (T7) and human cationic trypsinogen (PRSS1). The position of mutations used in the mouse models and the corresponding human mutations are indicated. Note that amino-acid numbering in mouse T7 trypsinogen is shifted by one relative to human PRSS1, e.g. Lys24 in T7 corresponds to Lys23 in PRSS1.
The p.D23A mutation increases autoactivation of mouse cationic trypsinogen dramatically, at least by 50-fold while activation by cathepsin B is unaffected. Phenotypically, the heterozygous T7D23A pups and the weaned mice look normal. They have slightly smaller bodyweight but they gain weight as well as C57BL/6N mice up to 5-6 months of age, after which their weight plateaus. They breed normally. Homozygous T7D23A mice are runted and have not been studied in detail.
Heterozygous T7D23A mice develop spontaneous, progressive pancreatitis at an early age (Figure 3). The pancreas of two-week-old mice looks normal but acute pancreatitis sets in typically at 3 weeks of age (range 3-5 weeks) with marked edema, high plasma amylase activity, massive inflammatory cell infiltration and acinar cell necrosis. Progression to chronic pancreatitis is fulminant and by 1-2 months of age essentially all mice exhibit pancreas atrophy with acinar cell dropout, diffuse fibrosis, pseudotubular complexes and persistent inflammatory cell infiltration. Further progression towards end-stage disease (6 months – 1 year) is characterized by large, dilated ducts, fatty infiltration that replaces fibrosis and apparent enlargement of islets. There is no overt diabetes, as judged by the blood glucose levels of 1-year-old mice. Acute pancreatitis can be observed in about a 30-35% of the mice sacrificed at 3-5 weeks of age, while the rest show chronic pathology. It is possible, even likely, that the acute stage is masked by the rapid progression in a large fraction of the mice and more frequent sampling would be required to capture this disease stage.
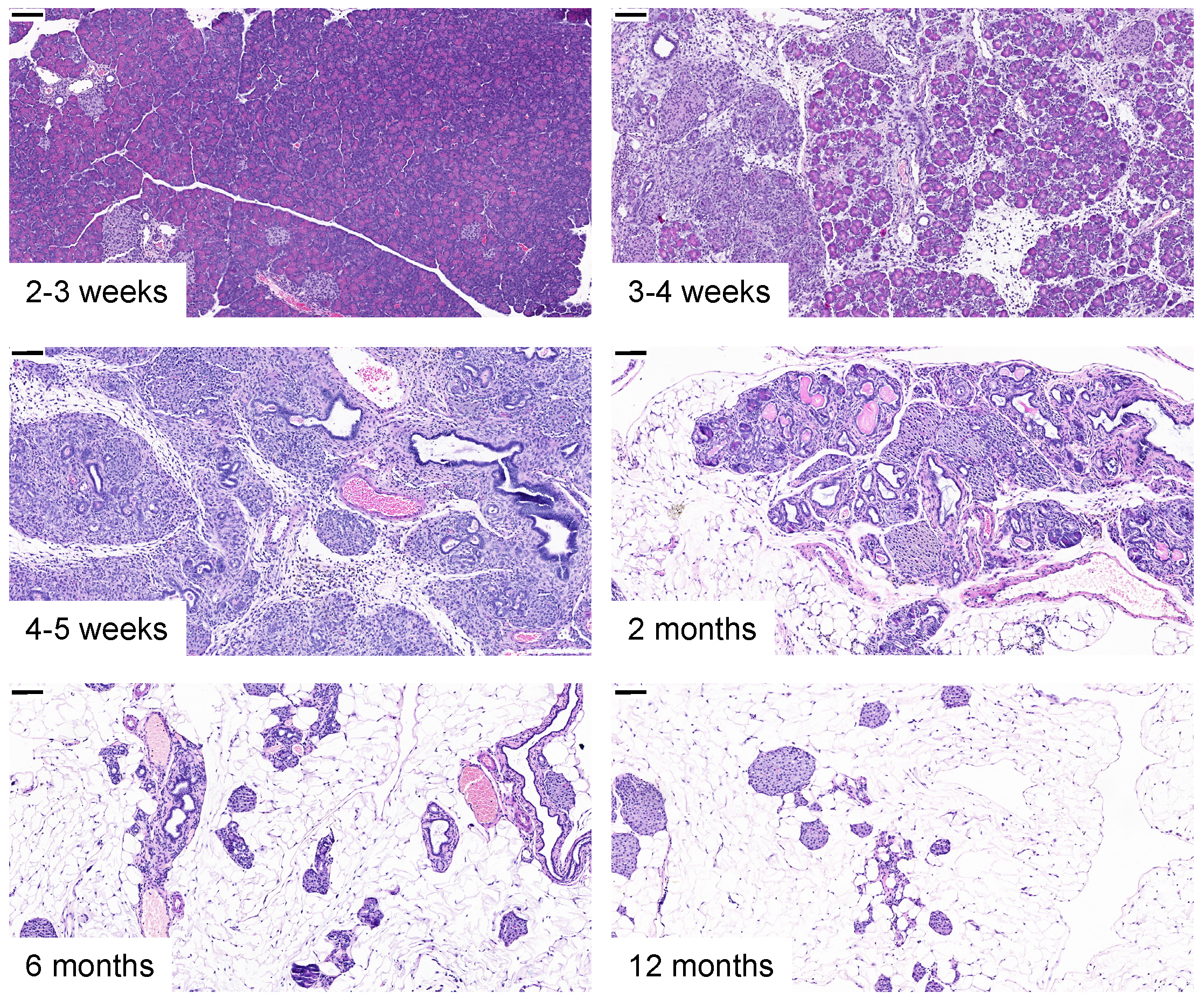
Figure 3. Spontaneous pancreatitis in T7D23A mice. Development of spontaneous acute pancreatitis and progression to chronic pancreatitis in T7D23A mice. Representative hematoxylin-eosin stained pancreas sections are shown. The scale bars correspond to 100 µM.
The T7D23A model was the first to demonstrate that increased trypsinogen autoactivation can cause spontaneous onset and progression of pancreatitis in mice. Furthermore, the model offered direct proof that acute and chronic pancreatitis are part of the same disease continuum driven by the same mechanism. As a preclinical model, the T7D23A strain is very promising. The mice breed well and exhibit no apparent symptoms related to their disease, with the exception of the lower bodyweight in older age. The disease develops rapidly and penetrance is 100%.
B. The T7K24R mouse model
Properties of cerulein-induced acute pancreatitis in these mice have been published in 2020 [14]. The T7K24R mice carry the p.K24R mutation in the activation peptide of T7 trypsinogen (Figure 2). This mutation corresponds to the p.K23R PRSS1 mutation found in human hereditary pancreatitis [6]. The mutation increases autoactivation of mouse cationic trypsinogen by about 5- fold and slightly reduces activation by cathepsin B. Heterozygous and homozygous T7K24R mice are phenotypically normal, they breed well, and develop no spontaneous pancreatic disease with the exception of scattered, focal lesions seen in 1-year-old mice. However, when given cerulein, T7K24R mice exhibit increased intrapancreatic trypsin activation and develop more severe acute pancreatitis, as judged by increased edema, higher plasma amylase activity and more robust inflammatory cell infiltration, relative to C57BL/6N mice (Figure 4).
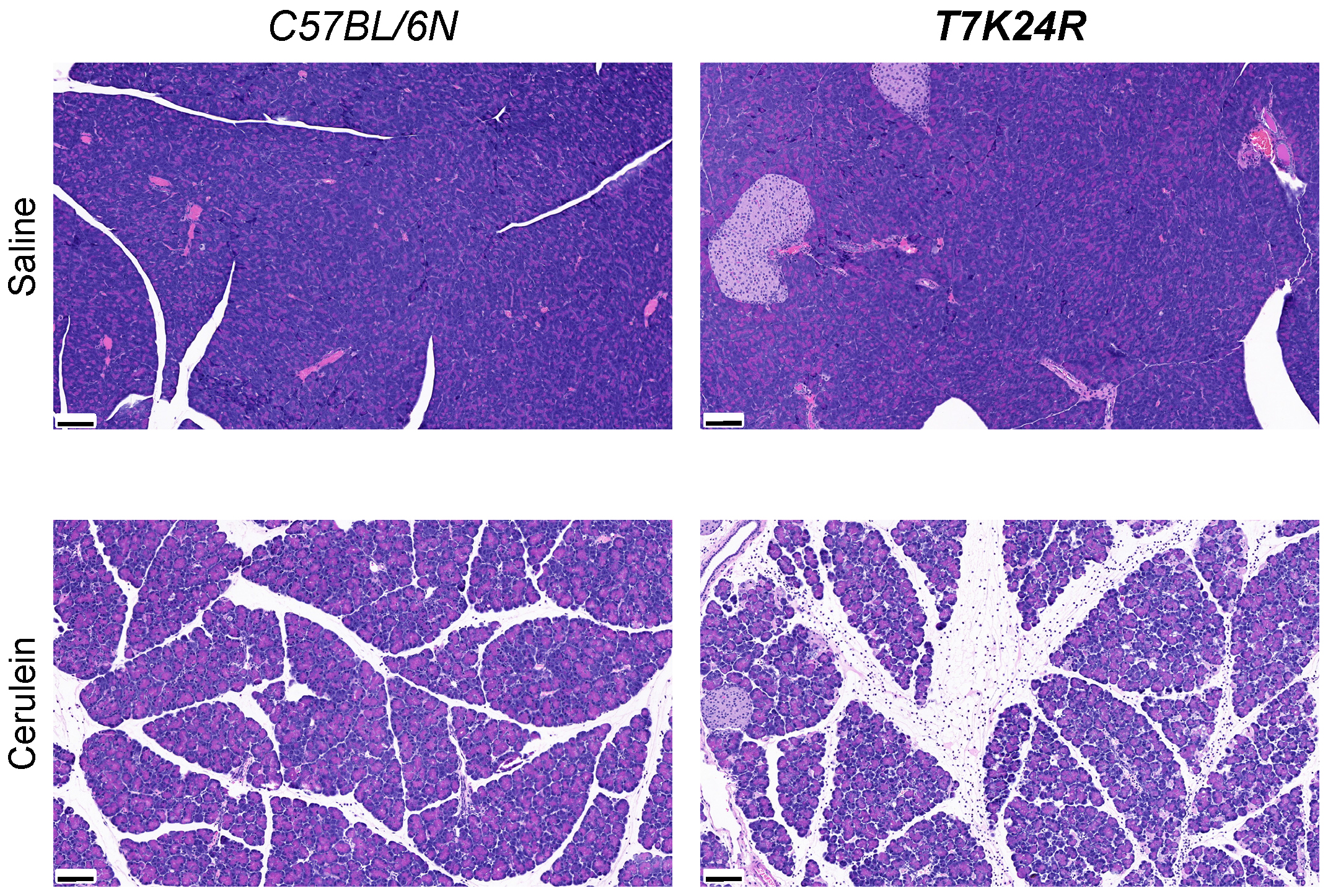
Figure 4. Cerulein-induced acute pancreatitis in T7K24R mice. Histology of cerulein-induced acute pancreatitis in T7K24R mice. Mice were treated with 12 hourly injections of saline or cerulein and sacrificed 1 h after the last injection. Representative hematoxylin-eosin stained pancreas sections are shown. The scale bars correspond to 100 µM.
Sustained stimulation of T7K24R mice with a 2-day regimen of cerulein injections resulted in a chronic pancreatitis-like disease phenotype, with acinar cell atrophy (dropout), pseudotubular complexes, inflammatory cells, and diffuse fibrosis, when mice were sacrificed 3 days or 10 days after the last injection (Figure 5). In contrast, similarly treated C57BL/6N mice rapidly recovered after two days of cerulein injections and showed only minimal changes in pancreas histology.
Taking the results together, we can conclude that the propensity of trypsinogen mutants for autoactivation determines pancreatitis responses and pathology. More robust autoactivation (T7D23A model) results in spontaneous pancreatitis while a smaller increase in autoactivation (T7K24R model) results in heightened sensitivity to experimentally induced pancreatitis.
As a preclinical model, T7K24R mice may be more useful than T7D23A mice when timing of pancreatitis onset needs to be well defined or when the extent of pancreatitis responses needs to be adjusted. For mechanistic studies in the academic laboratory setting, the T7K24R mice offer a clear advantage with robust pathological responses and easy-to-measure increases in inflammatory outcomes.
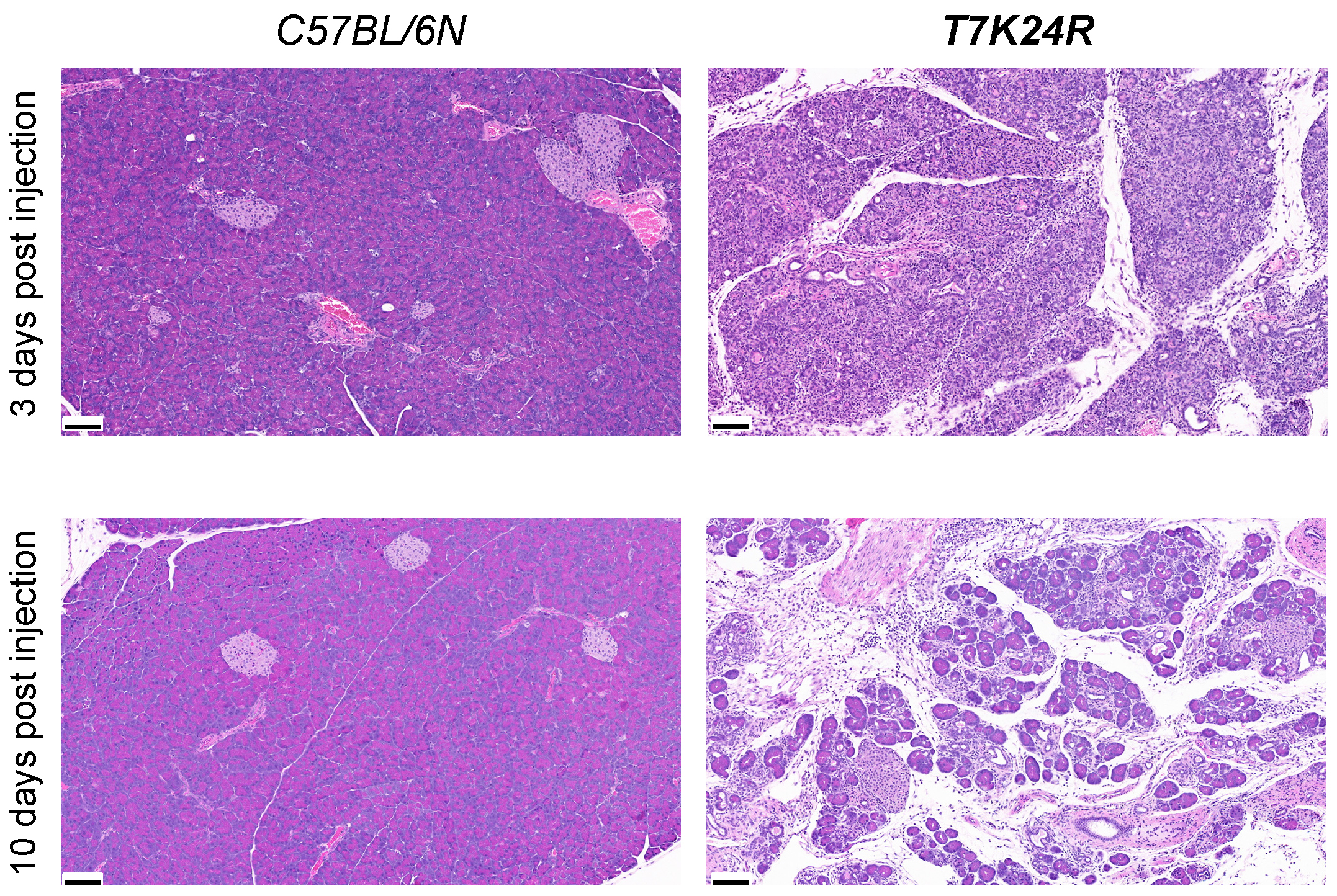
Figure 5. Cerulein-induced chronic pancreatitis in T7K24R mice. Histology of pancreatitis induced by sustained cerulein stimulation in T7K24R mice. Mice were treated with saline or cerulein for 2 days (8 hourly injections per day) and sacrificed 3 days or 10 days after the last injection. Representative hematoxylin-eosin stained pancreas sections are shown. The scale bars correspond to 100 µM.
II. Mouse Models Deficient in Chymotrypsin
In humans, CTRC plays a major protective role against pancreatitis by degrading trypsinogen and thereby mitigating intrapancreatic trypsin activation [3]. Chymotrypsin B1 and B2 (CTRB1, CTRB2) also contribute to protection to a much more limited extent, possibly through degradation of anionic trypsinogen (PRSS2) [15]. The role of chymotrypsin-like protease (CTRL) in pancreatitis has not been studied in humans so far. Mice express CTRB1, CTRC and CTRL, however, the C57BL/6 strains are naturally deficient in CTRC due to a single-nucleotide deletion in the Ctrc gene [16-18].
A. The Ctrb1-del model
Characteristics of the Ctrb1-del mouse model were reported in 2018 [16] and 2020 [17]. Using CRISPR-Cas9 genome editing, we deleted exon 4 of the Ctrb1 gene in mice. The resultant deletion allele expresses no detectable Ctrb1 mRNA or CTRB1 protein. Homozygous Ctrb1-del mice have a normal phenotype and develop no spontaneous pancreatitis. Total chymotrypsinogen content in the pancreas is reduced by 90% indicating that CTRB1 is the major isoform in mice. When given cerulein, Ctrb1-del mice exhibit higher intrapancreatic trypsin activation and more severe acute pancreatitis than C57BL/6N mice. The phenotype is highly similar to that of the T7K24R mice. Cerulein-induced intrapancreatic chymotrypsin activation is diminished by 75% in Ctrb1-del mice [17], indicating that CTRB1 is responsible for most of this activity. Taken together, properties of the Ctrb1-del mice indicate that chymotrypsin-deficiency increases sensitivity to cerulein-induced pancreatitis, in all likelihood due to loss of protective trypsinogen degradation and increased incrapancreatic trypsin activation. Indeed, biochemical experiments confirmed that CTRB1 degrades T7 trypsinogen and also inactivates the T8 and T9 isoforms by cleaving the autolysis loop [10, 16].
Besides its conceptual value, the Ctrb1-del strain is also important as a preclinical model of trypsin-dependent pancreatitis that contains no trypsinogen mutation. Furthermore, the mice may be crossed with trypsinogen mutant mice to study the interaction between chymotrypsin loss and increased trypsinogen activation.
B. The Ctrl-KO model
Properties of the Ctrl-KO mice were published in 2020 [17]. This strain was generated by CRISPR- Cas9 mediated genome editing, which was used to delete exon 3 of the Ctrl gene. The pancreas of Ctrl-KO mice expresses no Ctrl mRNA or CTRL protein. Homozygous Ctrl-KO mice exhibit a normal phenotype and do not develop spontaneous pancreatitis. Biochemically, mouse CTRL is capable of cleaving and inactivating T7, T8 and T9 trypsinogens, suggesting that it might participate in chymotrypsin-dependent defenses against pancreatitis. However, we found that CTRL protein expression levels in the mouse pancreas are low (10% or less of total chymotrypsinogen), which likely limits its contribution. When given cerulein, Ctrl-KO mice show a trend for increased intrapancreatic trypsin activation and slightly (by 25%) reduced chymotrypsin activation. Despite these changes in protease activation, severity of cerulein-induced pancreatitis remains similar to that of C57BL/6N mice. Overall, this phenotype suggests that CTRL plays no significant role in pancreatitis. The model has no direct preclinical value, but it may be crossed with Ctrb1-del to achieve complete elimination of chymotrypsin activity in C57BL/6N mice.
C. The C57BL/6N Ctrc+ model
Properties of this model were published in 2019 [18]. The C57BL/6 mouse strains are naturally deficient in CTRC. To investigate the protective role of CTRC in mice, we crossed the functional Ctrc locus from FVB mice to the C57BL/6N background through multiple generations. Biochemical analysis confirmed that mouse CTRC readily degrades T7 trypsinogen and inhibits autoactivation of T8 and T9 trypsinogens through cleavage of their autolysis loop [10]. When compared to mouse CTRB1, mouse CTRC had significantly more protective activity. Total chymotrypsinogen expression in the new Ctrc+ strain was elevated by 20% indicating that CTRC is a relatively minor isoform in mice. As expected, however, cerulein-induced intrapancreatic trypsin activity was reduced and severity of cerulein-induced pancreatitis was ameliorated in Ctrc+ mice relative to the C57BL/6N parent strain. This model provides important proof of concept for the protective role of CTRC against pancreatitis. The Ctrc+ mice can be crossed with various other strains in the C57BL/6N background to modify protective chymotrypsin levels and thereby moderate the pancreatitis phenotype.
III. Mouse Models Deficient in SPINK1
Deletion of mouse SPINK1 (legacy name SPINK3) was described in 2005 [19]. Homozygous mice were runted and died soon after birth, while heterozygous mice had no phenotype. Severity of cerulein-induced pancreatitis in heterozygous Spink1-KO mice was comparable to that of control mice [19, 20]. The pancreas of homozygous mice exhibited elevated trypsin activity, which is consistent with the lack of protective trypsin inhibition [21]. We recently generated a new Spink1- KO strain using CRISPR-Cas9 genome editing and characterization is underway.
To test the effect of SPINK1 deficiency on pancreatitis responses by an alternative method, we generated a mouse model carrying the human mesotrypsin signature mutation in mouse T7 trypsinogen. Properties of this model were published in 2021 [22]. Mesotrypsin is a minor human isoform resistant to trypsin inhibitors and capable of degrading SPINK1. The unique properties of mesotrypsin are mainly due to a single evolutionary mutation that replaced the conserved Gly198 residue with Arg. Mice homozygous for the corresponding p.G199R mutation in T7 trypsinogen had a normal phenotype and developed no spontaneous pancreatic disease. When given cerulein, intrapancreatic trypsin activation and severity of pancreatitis were similar in T7G199R and C57BL/6N mice. Importantly, no SPINK1 degradation was observed in the pancreas of cerulein-treated T7G199R mice. The observations suggest that human mesotrypsin, and mesotrypsin-mediated SPINK1 degradation in particular, do not play a significant role in pancreatitis development or severity.
IV. Mouse Models Overexpressing Trypsinogen
The first mouse models of trypsin-dependent pancreatitis were generated by introducing wild-type or mutant (p.N29I, p.R122H) forms of human PRSS1 or mouse anionic trypsinogen (isoform T8) as a transgene [23-25]. Although historically interesting, these models offered little useful insight and became obsolete. More recently, Dr. Baoan Ji developed and published a series of mouse models, some of which were generated in the laboratory of Dr. Craig Logsdon and some independently [26-30]. The mice carry transgenic rat or human trypsinogens and exhibit strong phenotypes of increased sensitivity to pancreatitis. These models will be reviewed elsewhere.
V. Mouse Model Overexpressing SPINK1
The protective function of SPINK1 against pancreatitis was also demonstrated by (over)expressing rat SPINK1 in the mouse pancreas from an elastase promoter [31-33].
VI. Future Perspectives
Recent years have seen the accelerated development of mouse models of trypsin-dependent pancreatitis. These new strains not only offer proof for the fundamental role of trypsin in pancreatitis pathogenesis but also serve as important preclinical models for therapeutic drug testing.
VII. References
- Whitcomb DC, Gorry MC, Preston RA, Furey W, Sossenheimer MJ, Ulrich CD, Martin SP, Gates LK Jr, Amann ST, Toskes PP, Liddle R, McGrath K, Uomo G, Post JC, Ehrlich GD. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet 1996, 14:141-145. PMID: 8841182.
- Witt H, Luck W, Hennies HC, Classen M, Kage A, Lass U, Landt O, Becker M. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet 2000, 25:213-216. PMID: 10835640.
- Rosendahl J, Witt H, Szmola R, Bhatia E, Ozsvári B, Landt O, Schulz HU, Gress TM, Pfützer R, Löhr M, Kovacs P, Blüher M, Stumvoll M, Choudhuri G, Hegyi P, te Morsche RH, Drenth JP, Truninger K, Macek M Jr, Puhl G, Witt U, Schmidt H, Büning C, Ockenga J, Kage A, Groneberg DA, Nickel R, Berg T, Wiedenmann B, Bödeker H, Keim V, Mössner J, Teich N, Sahin-Tóth M. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat Genet 2008, 40:78-82. PMID: 18059268.
- Hegyi E, Sahin-Tóth M. Genetic risk in chronic pancreatitis: The trypsin-dependent pathway. Dig Dis Sci 2017, 62:1692-1701. PMID: 28536777.
- Nemoda Z, Sahin-Tóth M. The tetra-aspartate motif in the activation peptide of human cationic trypsinogen is essential for autoactivation control but not for enteropeptidase recognition. J Biol Chem 2005, 280:29645-29652. PMID: 15970597.
- Férec C, Raguénès O, Salomon R, Roche C, Bernard JP, Guillot M, Quéré I, Faure C, Mercier B, Audrézet MP, Guillausseau PJ, Dupont C, Munnich A, Bignon JD, Le Bodic L. Mutations in the cationic trypsinogen gene and evidence for genetic heterogeneity in hereditary pancreatitis. J Med Genet 1999, 36:228-232. PMID: 10204851.
- Teich N, Ockenga J, Hoffmeister A, Manns M, Mössner J, Keim V. Chronic pancreatitis associated with an activation peptide mutation that facilitates trypsin activation. Gastroenterology 2000, 119:461-465. PMID: 10930381.
- Chen JM, Kukor Z, Le Maréchal C, Tóth M, Tsakiris L, Raguénès O, Férec C, Sahin-Tóth M. Evolution of trypsinogen activation peptides. Mol Biol Evol 2003, 20:1767-1777. PMID: 12832630.
- Yılmaz B, Ekiz F, Karakaş E, Aykut A, Simşek Z, Coban S, Onay H, Ozkinay F. A rare PRSS1 mutation in a Turkish family with hereditary chronic pancreatitis. Turk J Gastroenterol 2012, 23:826-827. PMID: 23864476.
- Németh BC, Wartmann T, Halangk W, Sahin-Tóth M. Autoactivation of mouse trypsinogens is regulated by chymotrypsin C via cleavage of the autolysis loop. J Biol Chem 2013, 288:24049- 24062. PMID: 23814066.
- Joergensen MT, Geisz A, Brusgaard K, Schaffalitzky de Muckadell OB, Hegyi P, Gerdes AM, Sahin-Tóth M. Intragenic duplication: a novel mutational mechanism in hereditary pancreatitis. Pancreas 2011, 40:540-546. PMID: 21499207.
- Geisz A, Hegyi P, Sahin-Tóth M. Robust autoactivation, chymotrypsin C independence and diminished secretion define a subset of hereditary pancreatitis-associated cationic trypsinogen mutants. FEBS J 2013, 280:2888-2899. PMID: 23601753.
- Geisz A, Sahin-Tóth M. A preclinical model of chronic pancreatitis driven by trypsinogen autoactivation. Nat Commun 2018, 9:5033. PMID: 30487519.
- Jancsó Z, Sahin-Tóth M. Mutation that promotes activation of trypsinogen increases severity of secretagogue-induced pancreatitis in mice. Gastroenterology 2020, 158:1083-1094. PMID: 31751559.
- Rosendahl J, Kirsten H, Hegyi E, Kovacs P, Weiss FU, Laumen H, Lichtner P, Ruffert C, Chen JM, Masson E, Beer S, Zimmer C, Seltsam K, Algül H, Bühler F, Bruno MJ, Bugert P, Burkhardt R, Cavestro GM, Cichoz-Lach H, Farré A, Frank J, Gambaro G, Gimpfl S, Grallert H, Griesmann H, Grützmann R, Hellerbrand C, Hegyi P, Hollenbach M, Iordache S, Jurkowska G, Keim V, Kiefer F, Krug S, Landt O, Leo MD, Lerch MM, Lévy P, Löffler M, Löhr M, Ludwig M, Macek M, Malats N, Malecka-Panas E, Malerba G, Mann K, Mayerle J, Mohr S, Te Morsche RHM, Motyka M, Mueller S, Müller T, Nöthen MM, Pedrazzoli S, Pereira SP, Peters A, Pfützer R, Real FX, Rebours V, Ridinger M, Rietschel M, Rösmann E, Saftoiu A, Schneider A, Schulz HU, Soranzo N, Soyka M, Simon P, Skipworth J, Stickel F, Strauch K, Stumvoll M, Testoni PA, Tönjes A, Werner L, Werner J, Wodarz N, Ziegler M, Masamune A, Mössner J, Férec C, Michl P, P H Drenth J, Witt H, Scholz M, Sahin-Tóth M; all members of the PanEuropean Working group on ACP. Genome- wide association study identifies inversion in the CTRB1-CTRB2 locus to modify risk for alcoholic and non-alcoholic chronic pancreatitis. Gut 2018, 67:1855-1863. PMID: 28754779.
- Jancsó Z, Hegyi E, Sahin-Tóth M. Chymotrypsin reduces the severity of secretagogue- induced pancreatitis in mice. Gastroenterology 2018, 155:1017-1021. PMID: 30076839.
- Mosztbacher D, Jancsó Z, Sahin-Tóth M. Loss of chymotrypsin-like protease (CTRL) alters intrapancreatic protease activation but not pancreatitis severity in mice. Sci Rep 2020, 10:11731. PMID: 32678161.
- Geisz A, Jancsó Z, Németh BC, Hegyi E, Sahin-Tóth M. Natural single-nucleotide deletion in chymotrypsinogen C gene increases severity of secretagogue-induced pancreatitis in C57BL/6 mice. J Clin Invest Insight 2019, 4:e129717. PMID: 31211695.
- Ohmuraya M, Hirota M, Araki M, Mizushima N, Matsui M, Mizumoto T, Haruna K, Kume S, Takeya M, Ogawa M, Araki K, Yamamura K. Autophagic cell death of pancreatic acinar cells in serine protease inhibitor Kazal type 3-deficient mice. Gastroenterology 2005,129:696-705. PMID: 16083722.
- Romac JM, Ohmuraya M, Bittner C, Majeed MF, Vigna SR, Que J, Fee BE, Wartmann T, Yamamura K, Liddle RA. Transgenic expression of pancreatic secretory trypsin inhibitor-1 rescues SPINK3-deficient mice and restores a normal pancreatic phenotype. Am J Physiol Gastrointest Liver Physiol 2010, 298:G518-524. PMID: 20110462.
- Ohmuraya M, Hirota M, Araki K, Baba H, Yamamura K. Enhanced trypsin activity in pancreatic acinar cells deficient for serine protease inhibitor kazal type 3. Pancreas 2006, 33:104-106. PMID: 16804421.
- Mosztbacher D, Sahin-Tóth M. Mouse model suggests limited role for human mesotrypsin in pancreatitis. Pancreatology 2021, 21:342-352. PMID: 33526384.
- Archer H, Jura N, Keller J, Jacobson M, Bar-Sagi D. A mouse model of hereditary pancreatitis generated by transgenic expression of R122H trypsinogen. Gastroenterology 2006, 131:1844- 1855. PMID: 17087933.
- Selig L, Sack U, Gaiser S, Klöppel G, Savkovic V, Mössner J, Keim V, Bödeker H. Characterisation of a transgenic mouse expressing R122H human cationic trypsinogen. BMC Gastroenterol 2006, 6:30. PMID: 17069643.
- Athwal T, Huang W, Mukherjee R, Latawiec D, Chvanov M, Clarke R, Smith K, Campbell F, Merriman C, Criddle D, Sutton R, Neoptolemos J, Vlatković N. Expression of human cationic trypsinogen (PRSS1) in murine acinar cells promotes pancreatitis and apoptotic cell death. Cell Death Dis 2014, 5:e1165. PMID: 24722290.
- Gaiser S, Daniluk J, Liu Y, Tsou L, Chu J, Lee W, Longnecker DS, Logsdon CD, Ji B. Intracellular activation of trypsinogen in transgenic mice induces acute but not chronic pancreatitis. Gut 2011, 60:1379-88. PMID: 21471572.
- ZhanX,WanJ,ZhangG,SongL,Gui F, Zhang Y, Li Y, Guo J, Dawra RK, Saluja AK, Haddock AN, Zhang L, Bi Y, Ji B. Elevated intracellular trypsin exacerbates acute pancreatitis and chronic pancreatitis in mice. Am J Physiol Gastrointest Liver Physiol 2019, 316:G816-G825. PMID: 30943050.
- Huang H, Swidnicka-Siergiejko AK, Daniluk J, Gaiser S, Yao Y, Peng L, Zhang Y, Liu Y, Dong M, Zhan X, Wang H, Bi Y, Li Z, Ji B, Logsdon CD. Transgenic expression of PRSS1R122H sensitizes mice to pancreatitis. Gastroenterology 2020, 1581072-1082. PMID:31419436.
- Gui F, Zhang Y, Wan J, Zhan X, Yao Y, Li Y, Haddock AN, Shi J, Guo J, Chen J, Zhu X, Edenfield BH, Zhuang L, Hu C, Wang Y, Mukhopadhyay D, Radisky ES, Zhang L, Lugea A, Pandol SJ, Bi Y, Ji B. Trypsin activity governs increased susceptibility to pancreatitis in mice expressing human PRSS1R122H. J Clin Invest 2020, 130:189-202. PMID: 31550238.
- Wan J, Haddock A, Edenfield B, Ji B, Bi Y. Transgenic expression of human PRSS2 exacerbates pancreatitis in mice. Gut 2020, 69:2051-2052. PMID: 31974135.
- Nathan JD, Romac J, Peng RY, Peyton M, Macdonald RJ, Liddle RA. Transgenic expression of pancreatic secretory trypsin inhibitor-I ameliorates secretagogue-induced pancreatitis in mice. Gastroenterology 2005, 128:717-727. PMID: 15765407.
- Nathan JD, Romac J, Peng RY, Peyton M, Rockey DC, Liddle RA. Protection against chronic pancreatitis and pancreatic fibrosis in mice overexpressing pancreatic secretory trypsin inhibitor. Pancreas 2010, 39:e24-30. PMID: 19904222.
- Romac JM, Shahid RA, Choi SS, Karaca GF, Westphalen CB, Wang TC, Liddle RA. Pancreatic secretory trypsin inhibitor I reduces the severity of chronic pancreatitis in mice overexpressing interleukin-1β in the pancreas. Am J Physiol Gastrointest Liver Physiol 2012, 302:G535-541. PMID: 22173919.