
Methods Type:
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2021.12
| Attachment | Size |
|---|---|
| 819.43 KB |
Pancreatitis is a common, potentially fatal disease of the exocrine pancreas. Its pathogenesis remains obscure, and no specific/effective treatment is available (28, 37, 39). Because of the general lack of access to human tissue, most studies use animal (as well as ex-vivo/cellular) models to investigate pathogenic mechanisms of pancreatitis (28). These models reproduce pathologic responses of human disease and the spectrum of its severity.
Autophagy is a principal degradative, lysosome-driven mechanism that eliminates damaged or unneeded cellular components and transports degradation products, such as amino acids and fatty acids, back to the cytoplasm to re-enter cellular metabolism (16, 17, 38). Autophagy mediates homeostatic functions of eukaryotic cells, acting as an intracellular organelle quality control system, and underlies adaptive cell responses to a variety of stress conditions (15, 25, 27, 35, 38, 50).
Impaired autophagy/lysosomal pathway is a critical feature of both human and experimental pancreatitis, and its role in pancreatitis is a matter of intense investigation (1, 5, 15-17, 22). Genetic models targeting autophagy/lysosomal pathways are an invaluable and widely used tool to examine the role of autophagy in cell physiology and various diseases (16, 17, 27 , 35, 38). Here we discuss utilization of autophagy-deficient genetic mouse models to better understand the role of autophagy in pancreatitis.
I. Autophagy/Lysosomal Pathway
A. Autophagy
Autophagy is a collective term for several pathways through which cytoplasmic material, particularly organelles and long-lived proteins, is delivered to the lysosome to be degraded by lysosomal hydrolases (25, 35, 38). The degradation serves two purposes: 1) removal of damaged or dysfunctional cellular components, such as uncoupled mitochondria or ubiquitylated protein aggregates, and 2) the provision of recycled substrates, such as amino acids and lipids, for critical cellular processes. The major (and best-studied) type of autophagy, macroautophagy, requires de novo formation of double-membraned structures, the autophagosomes, which sequester cargo and ultimately fuse with lysosomes to form autolysosomes, where degradation occurs. The process begins with the formation of so-called isolation membrane, or phagophore, followed by its elongation and closure to form the mature autophagosome. These steps are mediated by sequentially recruited complexes of evolutionary conserved ATG (autophagy-related) proteins (38, 50). In particular, autophagy initiation is controlled by ULK1/ATG1-mediated complex, followed by the formation of another multiprotein complex involving phosphatidylinositol 3-kinase catalytic subunit type 3 (Vps34) and Beclin1/ATG6, which nucleates the phagophore. Phagophore expansion and elongation are controlled by the ubiquitin-like conjugation systems involving the ATG5-ATG12-ATG16 complex and the microtubule-associated protein 1 light chain 3a (LC3). LC3, the mammalian paralog of yeast ATG8, is necessary for phagophore closure; during this process, its cytosolic form (LC3-I) is lipidated to become LC3-II, which translocates almost exclusively to the autophagosomal membranes (25, 27, 38, 50). Autophagosomes then fuse with lysosomes to form single-membraned autolysosomes where cargo breakdown occurs (Figure 1).
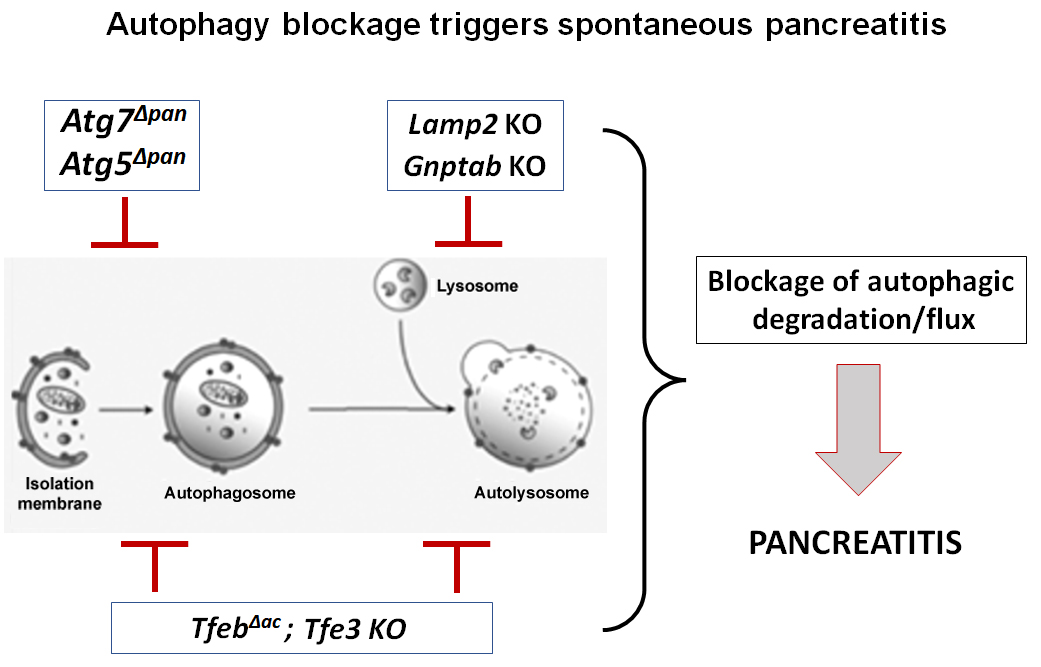
Figure 1. Blockage of autophagy/lysosomal pathway in genetically engineered mouse models causes pancreatitis.
B. Lysosomes
Lysosomes act as the waste disposal system of the cell; they degrade all kinds of unneeded biological material, including proteins, lipids and carbohydrates, coming from both inside and outside the cell (11, 41, 42). Material from outside the cell is taken up through endocytosis, while material from the inside of the cell is delivered to lysosomes through autophagy. Studies of the last decade have dramatically changed our view of the lysosome from that of a simple “garbage disposal” to a dynamic organelle that regulates basic cellular processes such as nutrient sensing, vesicular protein trafficking, endocytosis, and autophagy (11, 12, 41, 42).
The efficiency of lysosomal/autolysosomal degradation depends on lysosome’s degradative capacity, which requires several key elements. First, a unique mechanism for the sorting and delivery of a range of lysosomal enzymes to this compartment involves a covalent mannose 6-phosphate (M6P) addition to asparagine residues of many lysosomal (“acid”) hydrolases in the cis-Golgi network (8, 33, 42). N-acetylglucosamine-1-phosphotransferase is a key enzyme mediating the addition of M6P moieties onto acid hydrolases; its catalytic activity is mediated by α/β subunits coded by the GNPTAB gene (8, 33). The modified hydrolases then bind to one of the two transmembrane M6P receptors on vesicles that are subsequently directed toward endosomes to deliver their cargo to lysosomes.
The second characteristic feature of lysosomes is their acidic pH, which matches the pH optimum of most lysosomal enzymes and is modulated by a vacuolar H+-ATPase that mediates proton influx (15, 42). Lysosomal hydrolases, such as cathepsins, are often synthesized as pro-proteins and undergo proteolytic processing first in endosomes, then in lysosomes where acidic pH is required for their maturation to reach full catalytic activity (40). A third critical feature of lysosomes is the presence of essential membrane proteins, such as lysosome-associated membrane proteins LAMP1 and LAMP2 (11, 41, 42). LAMPs are transmembrane glycoproteins critical for maintaining lysosomal structure, its fusion with autophagosomes, and lysosomal proteolytic activity.
Recently discovered members of the MiT/TFE family of basic helix-loop-helix-zipper (bHLH-Zip) transcription factors (TFEB being the major one) regulate the expression of genes mediating lysosomal biogenesis and autophagy(42-45). In response to cellular stresses, TFEB, acting together with other MiT/TFE family members, such as TFE3, activates an efficient transcriptional program to control cellular degradation and facilitate intracellular clearance (43, 44).
II. Impairment of Autophagy/Lysosomal Pathway in Experimental and Human Pancreatitis
A. Lysosomal dysfunction in pancreatitis
Experimental pancreatitis is associated with severe defects in lysosomal functions (13, 15-18, 20, 21, 30, 32). In particular, pancreatic levels of LAMPs dramatically decrease in human disease and across various experimental models of nonalcoholic and alcoholic pancreatitis (13, 32). Pancreatitis also causes defects in the mechanisms mediating delivery of hydrolases to lysosomes. Both human and experimental pancreatitis cause a dramatic decrease in the level of GNPTAB (33). This is associated with defective processing/maturation of cathepsins, major lysosomal proteases, and with reduced cathepsins' enzymatic activities in lysosome-enriched pancreatic subcellular fractions, shown across rodent models of pancreatitis (6, 16, 30, 33).
TFEB and TFE3 are major MiT/TFE family members expressed in the pancreas. Both are active in healthy pancreas, but human and experimental pancreatitis exhibit a dramatic TFEB decrease in acinar cells indicating impaired autophagy and lysosomal biogenesis (47, 48).
B. Autophagy impairment in pancreatitis
Pancreatitis increases autophagosome formation in acinar cells; however, it inhibits autophagic degradation in autolysosomes due to defective lysosomal function, resulting in inhibition of autophagic flux (6, 15, 16, 30, 32, 33). Accumulation in acinar cells of cytoplasmic vacuoles, often filled with cellular debris, has long been recognized as an early marker of both human and experimental pancreatitis (2, 3, 7, 16, 24, 26, 36). Our studies showed that these vacuoles are autolysosomes containing partially degraded material and that their appearance is a result of the defective/inefficient autophagic degradation (6, 15, 16, 30, 32, 33). At the biochemical level, impaired autophagy is manifested in human and experimental pancreatitis by the concomitant increases in both the autophagic vacuole marker LC3-II and the autophagy substrate p62/SQSTM1, a sign of inefficient, retarded autophagic flux (6, 16, 25, 30, 32, 33, 48).
III. Genetic Models Utilized to Examine the Role of Autophagy/Lysosomal Pathway in the Pancreas
Several genetic models, targeting different steps in autophagy/lysosomal pathway, have been used toanalyze the effect of autophagy deficiency on pancreas (Table 1).

Table 1. Genetic models targeting autophagy/lysosomal pathway in pancreas.
Genetic ablation of Lamp2 or Gnptab severely impairs lysosomal proteolytic activity resulting in inhibition of the terminal step of autophagy, cargo degradation in autolysosomes (13, 32, 33). These models reproduce the decreases in pancreatic levels of LAMP2 and GNPTAB observed in human and experimental pancreatitis (13, 32, 33). Lamp2 ablation inhibits autophagic flux in pancreas and causes age-dependent development of pancreatitis, starting with acinar cell vacuolization and progressing to severe pancreas damage characterized by trypsinogen activation, macrophage-driven inflammation, and acinar cell death (32). Similarly, Gnptab ablation reduces lysosomal proteolytic activity in acinar cells, resulting in impaired autophagy and the development of spontaneous pancreatitis (33).
Ablation of Tfeb inhibits lysosomal biogenesis and autophagy at the transcriptional level, markedly aggravating experimental pancreatitis. Conversely, TFEB overexpression ameliorates the disease (47, 48). TFEB degradation is a prominent feature of both experimental and human pancreatitis; thus, its upregulation may be a promising strategy for preventing or treating pancreatitis. Tfeb ablation per se does not cause spontaneous pancreatitis development; however, silencing of both TFEB (specifically in acinar cells) and TFE3 causes severe autophagy/lysosomal impairment and spontaneous pancreatitis (47, 48). Of note, the functions of TFEB and TFE3 partially overlap, and TFE3 can compensate for the absence of TFEB (44, 45).
Inactivation of the key autophagy-related proteins ATG5or ATG7 (Table 1) blocks the first step in the process of autophagy, i.e., autophagosome formation (27, 38, 50). Pancreas specific ablation of either of them (Atg5Δpan and Atg7Δpan mice) causes the development of spontaneous pancreatitis with all the responses of human disease, i.e., inflammation, trypsinogen activation, fibrosis, acinar-to-ductal metaplasia, and pancreas atrophy (4, 9, 19). Of note, pancreatitis does not cause significant changes in the levels of ATG5 and ATG7 (31, 33). Thus, although Atg5Δpan and Atg7Δpan mice are a valuable tool to examine the role of autophagy in pancreatitis, these genetic models do not replicate initial events associated with pancreatitis.
The small GTPase Rab7 is a central regulator of membrane trafficking and plays key roles in the biogenesis of late endosomes and lysosomes, endocytosis, and autophagy progression. In particular, Rab7 is essential for autophagosome clustering in the perinuclear region and their subsequent fusion with lysosomes (14). Pancreas-specific Rab7 ablation causes impairment of autolysosome formation, evident by increased number of autophagosomes and reduction in autolysosomes in acinar cells, and exacerbates experimental acute pancreatitis (46).
LAMP2 and GNPTAB deficient acinar cells exhibit dysregulation of digestive enzyme secretion (32, 33), suggesting a role of autophagy in regulating the secretory function of exocrine pancreas. A possible underlying mechanism was suggested in a study (10) that showed the involvement of mediators of exocytosis in autophagosome formation. Specifically, the SNARE protein Syntaxin 2 (STX2) has been shown to inhibit both exocytosis and autophagy; the latter effect is due to STX2 interaction with the autophagy mediator ATG16L1. Genetic ablation of Stx2 both stimulates exocytosis and reduces the efficiency of autophagic flux, resulting in trypsinogen activation (10).
The genetic models targeting different steps in autophagy/lysosomal pathway (Table 1, #1-8) convincingly show that efficient basal autophagy plays a critical role in maintaining acinar cell homeostasis and its disruption by blocking either autophagosome formation or lysosomal function causes pancreas damage, resulting in pancreatitis. These findings are in marked contrast with those reported in an earlier study (23) in which ATG5 deficiency caused no damage in exocrine pancreas and even ameliorated some responses of cerulein-induced acute pancreatitis. A possible reason for this discrepancy may be the known suboptimal recombination efficiency of the Ela-Cre driver (29) used in that study.
All genetic models discussed above are in autophagy-deficient mice. By contrast, the overexpression of LC3 in GFP-LC3 transgenic mice (Table 1, #9) stimulates autophagosome formation. These mice were developed and then widely used to monitor autophagy in vivo, in different organs, as well as in isolated cells (25, 27, 34); they are particularly instrumental in measuring autophagy in organs with low autophagy rate. However, as shown in a recent study (31), pancreatic autophagy in GFP-LC3 mice is dysregulated, in that the increase in autophagosome formation is not balanced/matched by a corresponding increase in lysosomal degradation, resulting in reduced autophagic flux. LC3 overexpression in GFP-LC3 mice exacerbated experimental pancreatitis; most strikingly, it caused ~3-fold increases in hyperamylasemia, a diagnostic marker of acute pancreatitis, in several dissimilar mouse models (31). The results further underscore the importance of balanced, efficient autophagy for the exocrine pancreas; and indicate that application of GFP-LC3 mice should be done with caution, especially in disease models.
IV. Conclusions and Future Directions
The findings in genetic models convincingly demonstrate the essential role of autophagy/lysosomal pathway in maintaining acinar cell homeostasis and, further, that blockage or impairment of this pathway initiates and drives pancreatitis. Blockage of the first autophagy step, autophagosome formation, and the last step, cargo degradation, both cause the development of spontaneous pancreatitis. The data in these studies show that autophagy is critical for maintaining quality control of mitochondria and the endoplasmic reticulum (ER), the organelles essential for acinar cell survival and functions. Indeed, loss of autophagy in acinar cells causes accumulation of damaged mitochondria which fail to provide adequate ATP supply, and elicits ER and oxidative stress (4, 6, 9, 15, 18, 30, 32, 33, 48). Further, autophagy blockage in all the genetic models (Table 1) triggers acinar cell death and inflammation, indicating a strong pro-survival and anti-inflammatory role of autophagy in pancreas (4, 9, 15, 17, 20, 32, 33, 49, 51, 52).
Detailed analysis of the roles of autophagy in the exocrine pancreas has just begun, and we still know little about autophagy targets and underlying mechanisms. Genetic models targeting autophagy/lysosomal pathway represent an invaluable tool to advance our knowledge of how autophagy regulates pancreas physiology and pathophysiology. These models will be particularly useful in elucidating the mechanisms whereby autophagy/lysosomal pathway regulates digestive enzyme secretion and how disordering of this pathway leads to pancreatitis pathologies. Better understanding of the role of autophagy in pancreatitis responses, such as dysregulated secretion, inappropriate/intra-acinar activation of digestive enzymes, inflammation and cell death, could lead to new therapeutic targets and approaches to treat or mitigate the severity of pancreatitis. An important direction of future research is the development of genetic approaches to restore efficient autophagic degradation in pancreatitis (an attractive approach is upregulation of TFEB), and testing whether restoring efficient autophagy normalizes organellar (i.e., mitochondrial and ER) functions in acinar cells and ameliorates pancreatitis.
V. References
- Abu-El-Haija M, Gukovskaya AS, Andersen DK, Gardner TB, Hegyi P, Pandol SJ, Papachristou GI, Saluja AK, Singh VK, Uc A, and Wu BU. Accelerating the drug delivery pipeline for acute and chronic pancreatitis: summary of the working group on drug development and trials in acute pancreatitis at the National Institute of Diabetes and Digestive and Kidney Diseases workshop. Pancreas 47: 1185-1192, 2018. PMID: 30325856.
- Adler G, Rohr G, and Kern HF. Alteration of membrane fusion as a cause of acute pancreatitis in the rat. Dig Dis Sci 27: 993-1002, 1982. PMID: 7140496.
- Aho HJ, Nevalainen TJ, Havia VT, Heinonen RJ, and Aho AJ. Human acute pancreatitis: a light and electron microscopic study. Acta Pathol Microbiol Immunol Scand A 90: 367-373, 1982. PMID: 7148455.
- Antonucci L, Fagman JB, Kim JY, Todoric J, Gukovsky I, Mackey M, Ellisman MH, and Karin M. Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc Natl Acad Sci U S A 112: E6166-6174, 2015. PMID: 26512112.
- Barreto SG, Habtezion A, Gukovskaya A, Lugea A, Jeon C, Yadav D, Hegyi P, Venglovecz V, Sutton R, and Pandol SJ. Critical thresholds: key to unlocking the door to the prevention and specific treatments for acute pancreatitis. Gut 70: 194-203, 2021. PMID: 32973069.
- Biczo G, Vegh ET, Shalbueva N, Mareninova OA, Elperin J, Lotshaw E, Gretler S, Lugea A, Malla SR, Dawson D, Ruchala P, Whitelegge J, French SW, Wen L, Husain SZ, Gorelick FS, Hegyi P, Rakonczay Z, Jr., Gukovsky I, and Gukovskaya AS. Mitochondrial dysfunction, through impaired autophagy, leads to endoplasmic reticulum stress, deregulated lipid metabolism, and pancreatitis in animal models. Gastroenterology 154: 689-703, 2018. PMID: 29074451.
- Brackett KA, Crocket A, and Joffe SN. Ultrastructure of early development of acute pancreatitis in the rat. Dig Dis Sci 28: 74-84, 1983. PMID: 6822184.
- Coutinho MF, Prata MJ, and Alves S. Mannose-6-phosphate pathway: a review on its role in lysosomal function and dysfunction. Mol Genet Metab 105: 542-550, 2012. PMID: 22266136.
- Diakopoulos KN, Lesina M, Wormann S, Song L, Aichler M, Schild L, Artati A, Romisch-Margl W, Wartmann T, Fischer R, Kabiri Y, Zischka H, Halangk W, Demir IE, Pilsak C, Walch A, Mantzoros CS, Steiner JM, Erkan M, Schmid RM, et al. Impaired autophagy induces chronic atrophic pancreatitis in mice via sex- and nutrition-dependent processes. Gastroenterology 148: 626-638, 2015. PMID: 25497209.
- Dolai S, Liang T, Orabi AI, Holmyard D, Xie L, Greitzer-Antes D, Kang Y, Xie H, Javed TA, Lam PP, Rubin DC, Thorn P, and Gaisano HY. Pancreatitis-induced depletion of syntaxin 2 promotes autophagy and increases basolateral exocytosis. Gastroenterology 154: 1805-1821, 2018. PMID: 29360461.
- Eskelinen EL, Tanaka Y, and Saftig P. At the acidic edge: emerging functions for lysosomal membrane proteins. Trends Cell Biol 13: 137-145, 2003. PMID: 12628346.
- Ferguson SM. Beyond indigestion: emerging roles for lysosome-based signaling in human disease. Curr Opin Cell Biol 35: 59-68, 2015. PMID: 25950843.
- Fortunato F, Burgers H, Bergmann F, Rieger P, Buchler MW, Kroemer G, and Werner J. Impaired autolysosome formation correlates with Lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology 137: 350-360, 2009. PMID: 19362087.
- Guerra F, and Bucci C. Multiple roles of the small GTPase Rab7. Cells 5: 2016. PMID: 27548222.
- Gukovskaya AS, Gorelick F, Groblewski GE, Mareninova O, Lugea A, Antonucci L, Waldron RT, Habtezion A, Karin M, Pandol S, and Gukovsky I. Recent insights into the pathogenic mechanism of pancreatitis: role of acinar cell organelle disorders. Pancreas 48: 459-470, 2019. PMID: 30973461.
- Gukovskaya AS, and Gukovsky I. Autophagy and pancreatitis. Am J Physiol Gastrointest Liver Physiol 303: G993-G1003, 2012. PMID: 22961802.
- Gukovskaya AS, Gukovsky I, Algul H, and Habtezion A. Autophagy, inflammation, and immune dysfunction in the pathogenesis of pancreatitis. Gastroenterology 153: 1212-1226, 2017. PMID: 28918190.
- Gukovskaya AS, Pandol SJ, and Gukovsky I. New insights into the pathways initiating and driving pancreatitis. Curr Opin Gastroenterol 32: 429-435, 2016. PMID: 27428704.
- Gukovsky I, and Gukovskaya AS. Impaired autophagy triggers chronic pancreatitis: lessons from pancreas-specific atg5 knockout mice. Gastroenterology 148: 501-505, 2015. PMID: 25613315.
- Gukovsky I, Li N, Todoric J, Gukovskaya A, and Karin M. Inflammation, autophagy, and obesity: common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology 144: 1199-1209 e1194, 2013. PMID: 23622129.
- Gukovsky I, Pandol SJ, and Gukovskaya AS. Organellar dysfunction in the pathogenesis of pancreatitis. Antioxid Redox Signal 15: 2699-2710, 2011. PMID: 21834686.
- Habtezion A, Gukovskaya A, and Pandol S. Acute pancreatitis: a multi-faceted set of organellar, cellular and organ interactions. Gastroenterology 156: 1941-1950, 2019. PMID: 30660726.
- Hashimoto D, Ohmuraya M, Hirota M, Yamamoto A, Suyama K, Ida S, Okumura Y, Takahashi E, Kido H, Araki K, Baba H, Mizushima N, and Yamamura K. Involvement of autophagy in trypsinogen activation within the pancreatic acinar cells. J Cell Biol 181: 1065-1072, 2008. PMID: 18591426.
- Helin H, Mero M, Markkula H, and Helin M. Pancreatic acinar ultrastructure in human acute pancreatitis. Virchows Arch A Pathol Anat Histol 387: 259-270, 1980. PMID: 7456314.
- Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S, Abeliovich H, Abildgaard MH, Abudu YP, Acevedo-Arozena A, Adamopoulos IE, Adeli K, Adolph TE, Adornetto A, Aflaki E, Agam G, Agarwal A, Aggarwal BB, Agnello M, Agostinis P, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 17: 1-382, 2021. PMID: 33634751.
- Koike H, Steer ML, and Meldolesi J. Pancreatic effects of ethionine: blockade of exocytosis and appearance of crinophagy and autophagy precede cellular necrosis. Am J Physiol 242: G297-307, 1982. PMID: 7065251.
- Kuma A, Komatsu M, and Mizushima N. Autophagy-monitoring and autophagy-deficient mice. Autophagy 13: 1619-1628, 2017. PMID: 28820286.
- Lerch MM, and Gorelick FS. Models of acute and chronic pancreatitis. Gastroenterology 144: 1180-1193, 2013. PMID: 23622127.
- Magnuson MA, and Osipovich AB. Pancreas-specific Cre driver lines and considerations for their prudent use. Cell Metab 18: 9-20, 2013. PMID: 23823474.
- Mareninova OA, Hermann K, French SW, O'Konski MS, Pandol SJ, Webster P, Erickson AH, Katunuma N, Gorelick FS, Gukovsky I, and Gukovskaya AS. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J Clin Invest 119: 3340-3355, 2009. PMID: 19805911.
- Mareninova OA, Jia W, Gretler SR, Holthaus CL, Thomas DDH, Pimienta M, Dillon DL, Gukovskaya AS, Gukovsky I, and Groblewski GE. Transgenic expression of GFP-LC3 perturbs autophagy in exocrine pancreas and acute pancreatitis responses in mice. Autophagy 16: 2084-2097, 2020. PMID: 31942816.
- Mareninova OA, Sendler M, Malla SR, Yakubov I, French SW, Tokhtaeva E, Vagin O, Oorschot V, Lullmann-Rauch R, Blanz J, Dawson D, Klumperman J, Lerch MM, Mayerle J, Gukovsky I, and Gukovskaya AS. Lysosome associated membrane proteins maintain pancreatic acinar cell homeostasis: LAMP-2 deficient mice develop pancreatitis. Cell Mol Gastroenterol Hepatol 1: 678-694, 2015. PMID: 26693174.
- Mareninova OA, Vegh ET, Shalbueva N, Wightman CJ, Dillon DL, Malla S, Xie Y, Takahashi T, Rakonczay Z, Jr., French SW, Gaisano HY, Gorelick FS, Pandol SJ, Bensinger SJ, Davidson NO, Dawson DW, Gukovsky I, and Gukovskaya AS. Dysregulation of mannose-6-phosphate-dependent cholesterol homeostasis in acinar cells mediates pancreatitis. J Clin Invest 131: 2021. PMID: 34128834.
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, and Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 15: 1101-1111, 2004. PMID: 14699058.
- Morishita H, and Mizushima N. Diverse cellular roles of autophagy. Annu Rev Cell Dev Biol 35: 453-475, 2019. PMID: 31283377.
- Niederau C, and Grendell JH. Intracellular vacuoles in experimental acute pancreatitis in rats and mice are an acidified compartment. J Clin Invest 81: 229-236, 1988. PMID: 3335639.
- Pandol SJ, Saluja AK, Imrie CW, and Banks PA. Acute pancreatitis: bench to the bedside. Gastroenterology 132: 1127-1151, 2007. PMID: 17383433.
- Parzych KR, and Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 20: 460-473, 2014. PMID: 23725295.
- Peery AF, Crockett SD, Murphy CC, Lund JL, Dellon ES, Williams JL, Jensen ET, Shaheen NJ, Barritt AS, Lieber SR, Kochar B, Barnes EL, Fan YC, Pate V, Galanko J, Baron TH, and Sandler RS. Burden and cost of gastrointestinal, liver, and pancreatic diseases in the United States: Update 2018. Gastroenterology 156: 254-272 2019. PMID: 30315778.
- Rowan AD, Mason P, Mach L, and Mort JS. Rat procathepsin B. Proteolytic processing to the mature form in vitro. J Biol Chem 267: 15993-15999, 1992. PMID: 1639824.
- Saftig P, and Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol 10: 623-635, 2009. PMID: 19672277.
- Saftig P, and Puertollano R. How lysosomes sense, integrate, and cope with stress. Trends Biochem Sci 46: 97-112, 2021. PMID: 33012625.
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, and Ballabio A. TFEB links autophagy to lysosomal biogenesis. Science 332: 1429-1433, 2011. PMID: 21617040.
- Settembre C, Fraldi A, Medina DL, and Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 14: 283-296, 2013. PMID: 23609508.
- Steingrimsson E, Copeland NG, and Jenkins NA. Melanocytes and the microphthalmia transcription factor network. Annu Rev Genet 38: 365-411, 2004. PMID: 15568981.
- Takahashi K, Mashima H, Miura K, Maeda D, Goto A, Goto T, Sun-Wada GH, Wada Y, and Ohnishi H. Disruption of small GTPase Rab7 exacerbates the severity of acute pancreatitis in experimental mouse models. Sci Rep 7: 2817, 2017. PMID: 28588238.
- Wang S, Ni HM, Chao X, Ma X, Kolodecik T, De Lisle R, Ballabio A, Pacher P, and Ding WX. Critical role of TFEB-mediated lysosomal biogenesis in alcohol-induced pancreatitis in mice and humans. Cell Mol Gastroenterol Hepatol 10: 59-81, 2020. PMID: 31987928.
- Wang S, Ni HM, Chao X, Wang H, Bridges B, Kumer S, Schmitt T, Mareninova O, Gukovskaya A, De Lisle RC, Ballabio A, Pacher P, and Ding WX. Impaired TFEB-mediated lysosomal biogenesis promotes the development of pancreatitis in mice and is associated with human pancreatitis. Autophagy 15: 1954-1969, 2019. PMID: 30894069.
- Xue J, Sharma V, Hsieh MH, Chawla A, Murali R, Pandol SJ, and Habtezion A. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat Commun 6: 7158, 2015. PMID: 25981357.
- Zhao YG, and Zhang H. Autophagosome maturation: An epic journey from the ER to lysosomes. J Cell Biol 218: 757-770, 2019. PMID: 30578282.
- Zhong Z, Sanchez-Lopez E, and Karin M. Autophagy, inflammation, and immunity: A troika governing cancer and its treatment. Cell 166: 288-298, 2016. PMID: 27419869.
- Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D, Perkins G, Ali SR, McGeough MD, Ellisman MH, Seki E, Gustafsson AB, Hoffman HM, Diaz-Meco MT, Moscat J, and Karin M. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 164: 896-910, 2016. PMID: 26919428.