
Methods Type:
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2011.34
| Attachment | Size |
|---|---|
| 502.85 KB |
1. Background
Exocrine acinar cells have been the model of choice for examining phosphoinositide and Ca2+ signaling for decades. Initial evidence that muscarinic receptor activation resulted in PI metabolism was demonstrated by Hokin and Hokin using pancreatic slices in 1953 (6). Nearly ten years later, Douglas and Poisner established a requirement for Ca2+ in ACh-induced secretion from the structurally and functionally related submandibular salivary glands (3). While it was hypothesized that the two processes were linked (11), direct evidence for InsP3-induced mobilization of Ca2+ was not available until 1983 (16). In this landmark study, also using cells from the exocrine pancreas, Ca2+ was released from intracellular stores upon treatment of permeabilized cells with InsP3. Since then, this process has been documented in virtually every cell type and hundreds of different agonists have been shown to bind to receptors that trigger the production of InsP3 and the subsequent generation of Ca2+ signals (1).
Consistent with the polarized nature of acinar cells InsP3-mediated Ca2+ signaling in these cells exhibits highly organized spatial characteristics (15). Instead of Ca2+ rising uniformly in all regions of the cell, Ca2+ signals in acinar cells initiate in the apical region of the cells and proceed to the basolateral regions (4, 7). The basis for this organization likely arises from the subcellular localization of InsP3R (8). In fact, immunofluorescence studies have established that InsP3R are highly enriched in the apical domains of acinar cells (9, 13, 22). This specialized localization likely allows for very efficient activation of Ca2+ activated Cl- channels and zymogen granule fusion. The spread of Ca2+ signals to more distal regions of the cell allows information to be received by Ca2+ activated K+ channels and cell nuclei in the basal region of the cell (2, 10). At low levels of stimulation, Ca2+ signals can be retained in the apical domains (8, 19). Activation by levels of agonist thought to be in the physiological range typically produces repetitive Ca2+ transients, or oscillations (17, 23). This temporal regulation likely allows efficient activation of target molecules with the added advantage of preventing long-term elevations in [Ca2+]i that could be detrimental to the cell.
Acinar cells are, therefore, a well-established model system with which to study Ca2+ signaling. Acinar cells can be readily isolated from mammals while retaining functional and morphological features, thus allowing receptor-mediated Ca2+ signals to be observed in a native setting. Even after isolation, these cells produce highly organized receptor-induced Ca2+ signals that have been explicitly linked to physiologically important endpoints including zymogen granule exocytosis and Cl- channel activation (7, 14). Studies of Ca2+ signaling have also been useful in understanding processes involved in the etiology of pancreatitis. In particular, Ca2+ influx associated with depletion of the ER appears associated with the inappropriate intra-pancreatic activation of zymogens. Here, we describe methods to examine Ca2+ signals from various sources in isolated pancreatic acinar cells using wide-field digital imaging techniques.
2. Practical Considerations
2.1 Choice of calcium sensing dyes
A number of Ca2+ sensing dyes are available for use depending on the needs of the researcher (5, 12, 18). The simplest dyes are excited by a single wavelength of light and emit in a Ca2+ dependent manner at a single wavelength without a spectral shift. The prototypical dye of this class is Fluo3, which is excited by visible light with peak absorbance at 488 nm. Fluo3 and related dyes increase in fluorescence in response to Ca2+ binding. The equipment requirements for detection of single-wavelength dyes are relatively simple and because no wavelength switching is necessary these dyes are often the choice when rapid temporal information is required. While single wavelength dyes are obligate if one is limited to excitation from a laser source, these dyes do have some important drawbacks. Since the fluorescent signal is related to dye concentration, excitation strength and detector gain, these factors must all be kept constant throughout an experimental protocol.
The use of ratiometric Ca2+ sensing dyes obviates many of the concerns associated with single wavelength dyes. Fura2 is by far the most commonly used ratiometric dye for imaging Ca2+ signals in intact cells. Fura2 exhibits excitation spectrum changes upon Ca2+ binding such that the Ca2+ free form is excited maximally at 380 nm while the Ca2+ bound form is excited maximally at 340 nm. Both forms emit fluorescence with a peak at 510 nm allowing for a simple excitation ratio. This is in contrast to the other commonly used ratiometric dye Indo-1 which instead undergoes an emission shift upon Ca2+ binding necessitating two emission detectors or emission light splitters.
The development of modified versions of both single wavelength and ratiometric dyes has allowed for a library of dyes with various properties that can be exploited to measure Ca2+ in different domains. The most common variations are shifts in the affinity of the dyes for Ca2+ and excitation wavelengths. Dyes are available with affinities ranging from ~ 100 nM- 50 mM and thus can be used to monitor Ca2+ signals in various cellular compartments. The following provides detailed protocols for the measurement of cytosolic [Ca2+] from intact cells using either a ratiometric or single wavelength dye coupled with flash photolysis and further, a method for monitoring Ca2+ release from intracellular stores using a low affinity ratiometric probe.
3. Materials
3.1 Dyes
An array of fluorescent Ca2+ sensing probes are commercially available from companies such as Invitrogen and Teflabs. The most regularly used dyes for studies in pancreatic acinar cells are the AM forms of the Fura and Fluo classes. We prefer Fura2 for routine imaging and Fluo4 for single wavelength excitation applications. Varying excitation maximums ranging from 340 nm for Fura2 to 560 for Rhod2 allows multiplexing with other dyes and fluorescent markers.
3.2 Buffers
Imaging buffers consist of HEPES buffered saline solutions of the following composition:
| NaCl | 137 mM |
| KCl | 4.7 mM |
| NaH2PO4 |
1 mM |
| MgCl2 | 0.56 mM |
| CaCl2 | 1.8 mM |
| HEPES | 10 mM |
|
pH to 7.4 with NaOH |
|
4. Equipment
4.1 Microscope
We use either a Nikon TE200 Inverted microscope equipped with X 40 S Fluor objective (1.3 NA) or Olympus IX71 Inverted microscope with UAPO/340 objective (1.35 NA). Most commercial microscopes, whether upright or inverted coupled with high quality optics can be adapted for this use.
4.2 Light source
We use a Till Photonics Polychrome monochromator to provide rapidly switchable excitation light. The monochomator and image acquisition is controlled by TillVision software (till-photonics.com). Monochromaters based illuminators and integrated “Turnkey” imaging systems are also available from Cairn (Cairn-research.co.uk). Other researchers use halogen illumination coupled to appropriate filter changers to provide excitation light. Many solutions are available but popular filter based excitation systems are available from Sutter Instruments (Sutter.com) and Applied Scientific Imaging (Asiimaging.com) Laser illumination can also be used for single wavelength illumination.
4.3 Camera
In general imaging of cytosolic Ca2+ signals requires a camera with sufficient sensitivity for the detection of relatively low light levels, a reasonable resolution in terms of senor chip architecture, and the ability to collect images relatively rapidly. The weight placed on each of these characteristics should be matched to the Investigators requirements. For the majority of our applications, we use Cooke Sensicam QE 12 bit digital frame transfer cameras. The camera has a 12 bit dynamic range, is high resolution consisting of a sensor of 1376 x 1040 pixels with 6.45 μm pixel size (each pixel ~160 X 160 nm at 40 x), is peltier cooled (-14oC) which results in extremely low noise and is capable of imaging with exposure times of 500 nS. Less expensive, non cooled cameras with smaller format chips (i.e 512 x 512 pixels) are totally acceptable for more routine imaging, whereas cameras with back-thinned electron multiplication chips (EM cameras) may be required for rapid or low light level imaging. We have also used cameras from major manufacturers such as Hamamatsu (Hamamatsu.com), Andor (Andor.com) and Photometrics (photometrics.com) with equal success.
4.4 Imaging chamber
We typically use an Attofluor chamber from Invitrogen. An assortment of chambers are also available from Warner instruments.
4.5 Perfusion
Inverted syringes with luerlock valves provide convenient reservoirs which can form the basis of a gravity-fed perfusion system. We also use commercially purchased perfusion systems with electronically controlled pinch valves. These systems are available from Warner Instruments and Bioscience Tools.
5. Protocol for Imaging [Ca2+] in Pancreatic Acinar Cells
5.1 Cell isolation
High quality acinar cell preparations are essential for successful Ca2+ imaging experiments. Best results are typically obtained from small clusters (3-10 cells) of acinar cells. Cells are kept on ice and should be used within 6-8 hours following isolation. A protocol such as the one described by Williams in The Pancreapedia (20) should yield more than enough small clusters for a days worth of imaging.
5.2 Dye loading
a. Resuspend 50 μg of lyophilized Fura2-AM or Fluo4-AM with DMSO to yield a 1 mM stock. This stock should be kept in the dark at room temperature. A 1 mM stock solution of Fura-2 pentapotassium salt should be made for in vitro calibration standards.
b. Mix a 500 μL aliquot of acinar cells with Fura2-AM to a final concentration of 5 μM. Cell loading is performed in the dark to prevent degradation of the Fura2.
c. Incubate the cells with 1-5 μM Fura2 for 20 minutes at room temperature. Swirl the tube every 5-10 minutes to ensure consistent loading.
d. Centrifuge the cells at 500 g for 5 minutes and replace with HEPES imaging buffer without BSA.
5.3 Ca2+ imaging
a. Following loading, cells need to be mounted on a glass coverslip for imaging. Coverslips should be cleaned with 70% EtOH, rinsed with deionized water and thoroughly dried before mounting in the chamber. Any chamber which accepts a No 1 thickness coverslip will suffice.
b. Pipette 200-300 μL of acinar cells onto the coverslip. Cells will attach to the coverslip best if they are pipetted as a droplet on the center of the coverslip instead of being spread across the entire coverslip.
c. Allow cells to settle onto the coverslip for 10-15 minutes. This time also permits dye de-esterification.
d. Once the cells have settled, initiate superfusion of imaging buffer. A flow rate of ~ 3 ml/min is sufficient to allow rapid buffer exchange without disturbing cells that have attached to the coverslip. A multi-chamber gravity fed superfusion system is essential to perform multiple agonist applications within the course of the experiment. An electronically controlled device such as that produced by Warner instruments gives the added advantage of synchronizing agonist application with other instruments such as patch clamp amplifiers.
For experiments using Fura2, we typically alternately excite the dye loaded cells with 340 and 380 nm light at a rate of between 1-10 Hz and light emitted above 500 nM is collected by the camera. We routinely monitor “on line” the emission as a result of each excitation wavelength, as well as the 340/380 ratio during the experiment. A concurrent anti-parallel change in each signal should be reflected in the ratio change and is indicative of a change in [Ca2+] (Figure 1 A). Most commercial imaging software will calculate the mean ratio in multiple user defined region of interest (ROI) and published data is often simply reported as a change in the 340/380 ratio within a ROI without conversion to absolute [Ca2+] (Figure 1 B).Turnkey imaging systems typically are controlled by bespoke software, however several packages are available which have software drivers available to control a host of light sources and cameras. Examples include Metamorph (metamorph.com) and Imaging Workbench (Indecbiosystems.com). Increasingly drivers are also available for the free, NIH developed, imaging software “Image J” (rsbweb.nih.gov). Investigators are encouraged to explore this possibility and also to use this software for post acquisition analysis. Typical responses to physiological concentrations of the secretagogues carbachol and cholecystokinin are shown in figure 1 B/C and to a high concentration of CCK in 1D. Low concentrations of agonists tend to result in the initiation of an oscillating Ca2+ signal with specific characteristics depending on the stimulating agent. High concentrations uniformly result in a “peak and plateau” type response. Spatial information can also be gleaned from the images. A series of images depicting the apical initiation from a triplet of cells stimulated with 1 μM CCh is shown in Figure 2. Variations on the experimental conditions will allow the investigator to probe the contributions of various sources of Ca2+ using appropriate pharmacological tools and genetically modified mice. For example, a useful variation of this general paradigm is to assess the effects of removal of extracellular Ca2+ on the overall response as a means of gauging the contribution of Ca2+ influx to a particular response.

Figure 1. Ca2+ imaging with Fura2 in pancreatic acini. A. shows the kinetic traces from multiple ROIs either upon excitation with 340 nm light (upper panel) or 380 nm light (lower panel), emission was collected at 510 nm. Note the reciprocal anti-parallel change in the fluorescence signal which is diagnostic of a change in [Ca2+]i following stimulation with Carbachol. B. shows the 340/380 ratio change from the same ROIs generated from an image series where the 340 image is divided by the corresponding 380 image on a pixel-by-pixel basis. C. shows a kinetic of 340/380 ratio following stimulation with a low concentration of CCK. D. shows a kinetic from a cell stimulated with a maximal concentration of CCK.
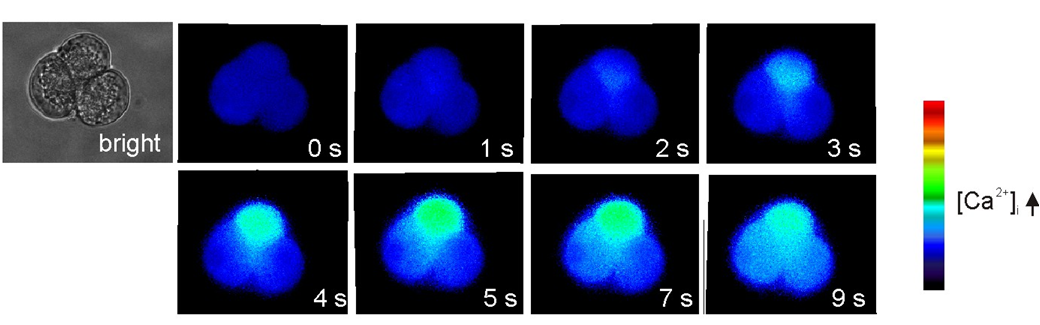
Figure 2. Spatial information from Fura2 imaging of pancreatic acini. A triplet of cells loaded with Fura2 was stimulated with 1 μM CCh prior to the image marked 0 s. The Ca2+ signal indicated by the change in color from dark blue, through light blue to green is initiated in the apical pole and spreads towards the basolateral face of the cell
5.4 Calibration of Fura-2 ratios to give an approximation of [Ca2+]i
Calibration of Fura-2 signals can be accomplished either in situ or in vitro. By far the simplest method is to calibrate the Fura2 signals in solution (in vitro) on the stage of the microscope. Effectively, Ca2+-EGTA solutions of known free [Ca2+] (range 10 nM- 20 mM) and pH are made in a buffer designed to mimic the intracellular milieu (125 mM KCl, 10 mM NaCl, 1 mM MgCl, 10 mM Hepes). The relative amounts of CaEGTA and EGTA necessary to generate a particular free [Ca2+] can be calculated using a program such as MaxChelator (available free from Maxchelator.stanford.edu). Alternatively, premade solutions are available as a Calcium Calibration Buffer Kit from Invitrogen. Fura2 free acid is then added to each solution to achieve a final concentration of 100 μM. A 50 μl droplet of each solution is then excited with 340 and 380 nm light and the ratio calculated. Care should be taken to ensure that identical exposure times, camera binning and plane of focus are used for each solution. We recommend etching the coverglass and focusing on the scratch to account for the latter consideration. The 340/380 ratio is then plotted vs. the [Ca2+] and the ratios obtained from acini can be compared with the standard curve generated. This method of calibration using a simple protocol yields an approximation of cellular [Ca2+]. However, because the behavior of indicator dyes can be markedly affected by factors such as osmolality, pH, ionic strength, viscosity and the intracellular protein compliment it is estimated that the calibration in solution introduces as much as 15% error to the calculation of [Ca2+]i. Therefore, the “gold standard” is to perform the calibration in situ.
In its simplest form a two-point calibration to determine 340/380 ratios at minimum Ca2+ and saturating Ca2+ is performed. An intracellular type buffer containing either 10 mM Ca2+ or 1 mM EGTA at pH 7.4 with 10 μM Ca2+ ionophore, (Ionomycin or 8Br-A23187) is prepared as the MAX and MIN Ca2+ buffer, respectively. Seed Fura-2-AM loaded acini onto coverglass as previously described. The acini are then perfused with the MIN buffer until a new steady state 340/380 ratio is obtained (5-10 mins). Subsequently, the solution is exchanged with the MAX buffer, again until a new steady state 340/380 ratio is obtained. These maneuvers yield values which can be input to the Grynkiewicz equation (5) used to calculate intracellular free Ca2+.
For eg.
Where Kd is Fura-2 effective dissociation constant (estimated at 135 or 225 nM at 22 or 37oC respectively).
Fmin380 is the fluorescence intensity following excitation at 380 nM in MIN solution.
Fmax380 is the fluorescence intensity following excitation at 380 nM in MAX solution.
Rmin is the 340/380 ratio in MIN solution.
Rmax is the 340/380 ratio in MAX solution.
Using these values the investigator can use this equation to solve for R, the observed ratio following an experimental intervention.
Additional Considerations: In our experience it can be difficult to obtain stable values representing the ratios for free and fully bound indicator as acini attempt to regulate [Ca2+]i despite the presence of the ionophore. We have found that depleting ATP to inhibit ATPases can ameliorate this issue. Practically this can be achieved by incubating cells in 10 μM rotenone combined with replacing glucose for deoxy-D-glucose 10 mins prior to the calibration. The above 2-point in situ calibration can be also extended using similar experimental procedures but utilizing a range of Ca2+ concentrations (see (18) for discussion of this technique).
6. Protocol for Measuring [Ca2+]i Changes Following Photolysis of Caged Molecules in Intact Acini
A powerful variation of this general imaging paradigm described above is to combine the image acquisition with a means to provide concurrent illumination with a UV light source to photolyse chemically caged second-messenger precursors. Currently, cell-permeable caged versions of Ca2+ and InsP3 are available from Invitrogen and Axxora, respectively. These compounds can be loaded into cells in a similar fashion to the indicator dyes as a function of lipophillic cleavable ester moieties conjugated to the cage. Because photo-destruction of the cage is accomplished by UV light, imaging is usually performed utilizing single wavelength dyes excited by visible light to ensure that image acquisition per se does not release active second-messenger. Slight modifications to the protocol and equipment described in section 5 are needed to perform these experiments.
6.1 Equipment
All equipment described in section 4 is appropriate for these experiments. In addition a suitable UV light source together with a means to integrate this to the imaging system is necessary. Suitable light sources include UV flash lamps and lasers either focused through the epifluorescence condenser or focused from above the stage. We use a condenser which allows simultaneous fiber optic input from the Till monochromator and UV light source directed to the objective through a 400 nm dichroic mirror. This arrangement separates the imaging light from the photolysis light and provides a brief (50 mS) pulse of intense UV light across the entire imaging field. This equipment is available from Till Photonics. Cairn Instruments and Rapp Optoelectronics also sells elegant solutions to provide UV light for flash photolysis.
6.2 Dye loading
Repeat steps 5.2 a-d incubating cells with 1-5 μM Fluo4-AM and 1-5 μM Caged InsP3-PM or 10-50 μM NP-EGTA-AM (caged Ca2+) concurrently.
6.3 Imaging
Repeat steps 5a-d.
Fluo4 is excited at 488 nm and emission is detected above 520 nm. More rapid data acquisition can be achieved using single wavelength indicators (~ 50 Hz with our imaging system). Typically we image for a period to establish a baseline and then trigger the UV pulse (~50-100 ms exposure at user defined energy level). Varying energy allows a grading of the intensity of UV exposure and in turn active second-messenger released. This can be performed manually or controlled by analog/digital input pulse and controlled by computer software. A series of images and the kinetic plots of a typical experiment where InsP3 is released locally in the apical pole of a pancreatic acini is shown in Figure 3. Data is often presented as a pseudo-ratio where each image is normalized to an average of some number of baseline images. We typically will present images which are the change in fluorescence (ΔF) is divided by the mean of the initial 10 frames (F0) on a frame-by-frame basis. This analysis facilitates comparing changes in fluorescence between individual cells.
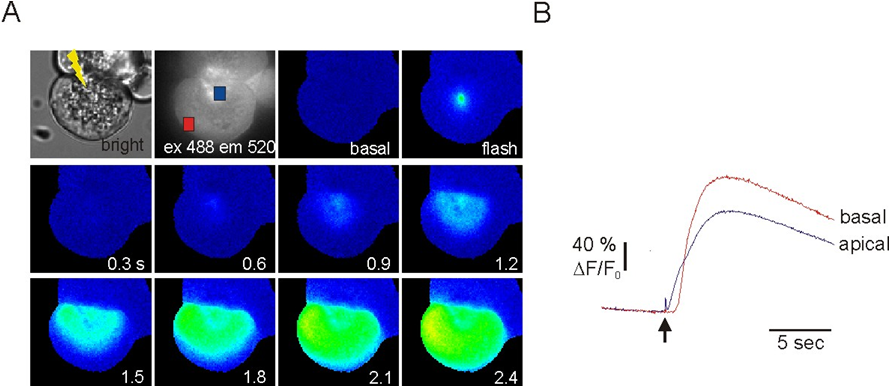
Figure 3. Flash photolysis of caged-IP3 with simultaneous imaging of Fluo4 in pancreatic acini. In A, IP3 is released in the location indicated by the “lightning bolt” in the apical pole shown in the bright field image. ΔF/F0 Images of the resultant Ca2+ signal are shown at the indicated times. The signal initiated in the apical pole and spreads towards the basal pole. B, shows the kinetic of the change in fluorescence following uncaging in the apical and basal pole. Data is modified from (21).
7. Protocol for Measuring Ca2+ Release from Intracellular Stores
The protocols described above are suitable for recording Ca2+ responses to secretagogues and second messengers in intact cells. Given the importance of various intracellular stores in initiating and shaping Ca2+ signals, there is much interest in probing the regulation of the channels and pumps that comprise the “calcium handling toolkit” of the pancreatic acinar cell. However, the intracellular localization of important channels such as IP3R generally precludes direct measurement by patch clamp methodology typically used to record ion channel activity. Selective permeabilization of the plasma membrane allows access to intracellular stores and facilitates tight control intracellular constituents. The protocol below describes a typical application of permeabilized preparation to analyze the regulation of pancreatic IP3R function.
7.1 Experimental buffers
Careful buffer preparation is essential to obtain meaningful information. Ensure the proper pH of all experimental solutions since the affinity of EGTA for Ca2+ is highly pH-dependent. The free [Ca2+] in buffers should also be verified using fluorimetry measurements with Fura2 or with a Ca2+ electrode.
7.2 Ca2+ dye loading
Pancreatic acinar cells for this application can be isolated by the standard method and resuspended in HEPES-PSS supplemented with 1% BSA containing 10 μM of Furaptra-AM (MagFura) a low affinity Ca2+ indicator. For this application, cells are loaded with Furaptra-AM at 37°C, 5% CO2 for 30 minutes to facilitate sequestration of the dye in the ER lumen. After loading is complete, cells can be mounted on a coverlslip at the bottom of a small-volume perfusion chamber as described for intact cell imaging.
7.3 Pancreatic acinar cell permeabilization
a. Permeabilize the cells by perfusing with a buffer solution that contains ions at the expected intracellular concentrations (ICM: 125 mM KCl, 10 mM NaCl, 1 mM EGTA, 10 mM HEPES, pH 7.3) and containing 40 μM β-escin.
b. Monitor the dye leaving the cytosol to allow proper timing of the permeabilization process. If β-escin is allowed to permeabilize intracellular membranes it becomes increasing difficult to load Ca2+ into the ER lumen.
c. Excite cells with 360 nm light which is the isosbestic point for Furaptra at 0.2 Hz intervals. An example of pancreatic acinar cell permeabilization is shown in figure 4A/B. Cells loaded with Furaptra prior to β-escin treatment are shown in figure 4a.
d. Initiate permeabilization by superfusion with Ca2+ free ICM containing 40 μM β-escin and wash the β-escin off when the fluorescence falls below 20% of the original fluorescence (Figure 4B). This process typically takes one to two minutes as shown in Figure B. Cells after permeabilization are shown in Figure 4Ab. After permeabilization, continue to wash cells with Ca2+-free ICM for 15-20 minutes. This allows passive depletion of Ca2+ from ER stores.

Figure 4. InsP3-induced Ca2+ release from permeabilized pancreatic acinar cells. A shows fluorescence images an acinus loaded with Furaptra at excited with 360 nm light, prior to (Aa) and post (Ab) permeabilization. Images Ac-e are 340/380 nm ratio images captured at the time points indicated in B. In B cells were permeabilized with β-escin to allow removal of cytoplasmic dye (left panel) and incubated in Ca2+ free internal solution for 20 minutes. The right panel of B shows a typical load and release protocol. Ca2+, Mg2+ and ATP were added at the indicated time and the increase in 340/380 ratio indicates Ca2+ entering the lumen of the ER. Mg2+ was removed as indicated to inactivate SERCA and the subsequent application 10 μM InsP3 empties the stores as indicated by the decreasing 340/380 ratio. C. shows normalized representative responses to the indicated concentrations of InsP3. D. shows the average Ca2+ release rates in response to a range of InsP3 concentrations.
7.4 Loading ER stores with Ca2+
a. Load the ER stores by superfusion with ICM supplemented with 1.4 mM MgCl2, 3 mM Na2ATP and 650 μM CaCl2 (2 are calculated to be 1.3 mM and 200 nM respectively. These conditions allow for optimal activation of SERCA.
b. Monitor the 340/380 ratio to ensure an increase in fluorescence ratio which indicates Ca2+ entering the lumen of the ER. ER loading is typically complete within 2-3 minutes as indicated by a stabilization of the 340/380 ratio (Figure 4B right panel).
c. Remove MgCl2 from the perfusion buffer for one minute prior to InsP3 application. This effectively eliminates SERCA activity, which allows the measurement of unidirectional Ca2+ release through IP3R.
7.5 InsP3-induced Ca2+ release from ER stores
a. Apply ICM buffer containing desired Ca2+, ATP and InsP3 concentrations. There will be a resulting decrease in the 340/380 ratio with [InsP3]. During the “releasing” phase of the experiment, acquire images at 1-2 Hz. A major benefit to this protocol is that multiple Ca2+ release events can be monitored from the same batch of cells. Also, recordings from multiple cells can be made simultaneously and averaged together.
b. Rapid exchange of buffer solutions is needed to obtain kinetic information about the rate of Ca2+ release. For the experiments described in figure 4, flow rates were typically greater than 4 ml/minute providing rapid exchange of the small volumes (< 500 μL) present in the imaging chamber.
c. In order to mitigate bleaching, use higher binning and reduce the exposure times (4 x 4 and 20-40 ms respectively) during image acquisition. In addition, limit acquisition rates to 0.1 Hz during the “loading” phase when kinetic information is not required. Even under optimal conditions, bleaching typically prevents recording of more than three or four release events per experiment.
7.6 Data processing
Relative InsP3R activity under various experimental conditions can be determined from these recordings by fitting the decrease in fluorescence ratios to exponential functions. Limiting the fitting to the initial 20-30% reduction in 340/380 ratio allows fits to be made with single exponentials. An example of the type of information gained using these conditions is shown in figure 4C and D.
8. References
- Berridge, M. J., P. Lipp, and M. D. Bootman. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1: 11-21, 2000. PMID: 11413485
- Bruce, J. I., T. J. Shuttleworth, D. R. Giovannucci, and D. I. Yule. Phosphorylation of inositol 1,4,5-trisphosphate receptors in parotid acinar cells. A mechanism for the synergistic effects of cAMP on Ca2+ signaling. J Biol Chem 277: 1340-1348, 2002. PMID: 11694504
- Douglas, W. W. and A. M. Poisner. The influence of calcium on the secretory response of the submaxillary gland to acetylcholine or to noradrenaline. J Physiol 165: 528-541, 1963. PMID: 16992144
- Giovannucci, D. R., J. I. Bruce, S. V. Straub, J. Arreola, J. Sneyd, T. J. Shuttleworth, and D. I. Yule. Cytosolic Ca(2+) and Ca(2+)-activated Cl(-) current dynamics: insights from two functionally distinct mouse exocrine cells. J Physiol 540: 469-484, 2002. PMID: 11956337
- Grynkiewicz, G., M. Poenie, and R. Y. Tsien. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260: 3440-3450, 1985. PMID: 3838314
- Hokin, M. R. and L. E. Hokin. Enzyme secretion and the incorporation of P32 into phospholipides of pancreas slices. J Biol Chem 203: 967-977, 1953. PMID: 13084667
- Kasai, H. and G. J. Augustine. Cytosolic Ca2+ gradients triggering unidirectional fluid secretion from exocrine pancreas. Nature 348: 735-738, 1990. PMID: 1701852
- Kasai, H., Y. X. Li, and Y. Miyashita. Subcellular distribution of Ca2+ release channels underlying Ca2+ waves and oscillations in exocrine pancreas. Cell 74: 669-677, 1993. PMID: 8395348
- Lee, M. G., X. Xu, W. Zeng, J. Diaz, R. J. Wojcikiewicz, T. H. Kuo, F. Wuytack, L. Racymaekers, and S. Muallem. Polarized expression of Ca2+ channels in pancreatic and salivary gland cells. Correlation with initiation and propagation of [Ca2+]i waves. J Biol Chem 272: 15765-15770, 1997. PMID: 9188472
- Maruyama, Y., D. V. Gallacher, and O. H. Petersen. Voltage and Ca2+-activated K+ channel in baso-lateral acinar cell membranes of mammalian salivary glands. Nature 302: 827-829, 1983. PMID: 6302513
- Michell, R. H. Inositol phospholipids and cell surface receptor function. Biochim Biophys Acta 415: 81-47, 1975. PMID: 164246
- Minta, A., J. P. Kao, and R. Y. Tsien. Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores. J Biol Chem 264: 8171-8178, 1989. PMID: 2498308
- Nathanson, M. H., M. B. Fallon, P. J. Padfield, and A. R. Maranto. Localization of the type 3 inositol 1,4,5-trisphosphate receptor in the Ca2+ wave trigger zone of pancreatic acinar cells. J Biol Chem 269: 4693-4696, 1994. PMID: 7508924
- Park, M. K., R. B. Lomax, A. V. Tepikin, and O. H. Petersen. Local uncaging of caged Ca(2+) reveals distribution of Ca(2+)-activated Cl(-) channels in pancreatic acinar cells. Proc Natl Acad Sci U S A 98: 10948-10953, 2001. PMID: 11535807
- Petersen, O. H. and A. V. Tepikin. Polarized calcium signaling in exocrine gland cells. Annu Rev Physiol 70: 273-299, 2008. PMID: 17850212
- Streb, H., R. F. Irvine, M. J. Berridge, and I. Schulz. Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature 306: 67-69, 1983. PMID: 6605482
- Stuenkel, E. L., Y. Tsunoda, and J. A. Williams. Secretagogue induced calcium mobilization in single pancreatic acinar cells. Biochem Biophys Res Commun 158: 863-869, 1989. PMID: 2920043
- Thomas, D., S. C. Tovey, T. J. Collins, M. D. Bootman, M. J. Berridge, and P. Lipp. A comparison of fluorescent Ca2+ indicator properties and their use in measuring elementary and global Ca2+ signals. Cell Calcium 28: 213-223, 2000. PMID: 11032777
- Thorn, P., A. M. Lawrie, P. M. Smith, D. V. Gallacher, and O. H. Petersen. Local and global cytosolic Ca2+ oscillations in exocrine cells evoked by agonists and inositol trisphosphate. Cell 74: 661-668, 1993. PMID: 8395347
- Williams, J. A. Isolation of rodent pancreatic acinar cells and acini by collagenase digestion. The Pancreapedia: Exocrine Pancreas Knowledge Base DOI: 10.3998/panc.2010.18, 2010.
- Won, J. H., W. J. Cottrell, T. H. Foster, and D. I. Yule. Ca2+ release dynamics in parotid and pancreatic exocrine acinar cells evoked by spatially limited flash photolysis. Am J Physiol Gastrointest Liver Physiol 293: G1166-1177, 2007. PMID: 17901163
- Yule, D. I., S. A. Ernst, H. Ohnishi, and R. J. Wojcikiewicz. Evidence that zymogen granules are not a physiologically relevant calcium pool. Defining the distribution of inositol 1,4,5-trisphosphate receptors in pancreatic acinar cells. J Biol Chem 272: 9093-9098, 1997. PMID: 9083036
- Yule, D. I. and D. V. Gallacher. Oscillations of cytosolic calcium in single pancreatic acinar cells stimulated by acetylcholine. FEBS Lett 239: 358-362, 1988. PMID: 3141216