
Methods Type:
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2022.02
| Attachment | Size |
|---|---|
| 1.34 MB |
I. Introduction
Pancreatic cancer continues to be a significant cause of mortality across the world. Currently, it is the 3rd leading cause of cancer death in the United States, and it is often diagnosed late when there are few treatment options. The 5-year survival rate remains dismal at approximately 10% (72). Patients frequently endure severe abdominal pain, jaundice, ascites, and cachexia. A subset of these patients will also develop diabetes mellitus. Acinar cells are the prevalent cell type of the pancreas, and a large portion of human pancreatic carcinomas are pancreatic ductal adenocarcinomas. Acinar cells are able to dedifferentiate through a process known as acino-ductal metaplasia, and some believe that the resulting duct like structures result in pancreatic ductal adenocarcinoma.
It is essential to note that mutations in kras and p53, commonly found in human pancreatic cancer, are also found in chemical-induced models of pancreatic cancer. A large majority of pancreatic ductal adenocarcinomas occur in the head of the pancreas, and these are known to have strong metastatic potential. This review will focus on chemically induced animal models of pancreatic cancer in different species, elucidating the typical timeframe to the development of pancreatic cancer, comparing histologic and molecular features to pancreatic cancer in humans, and the other off-target cancers that may result.
II. Nitrosamines
Nitrosamines are organic compounds with carcinogenic potential that have been extensively studied. Herein, we will review different nitrosamine compounds by their composition and the resulting carcinogenesis.
A. N-nitrosobis (2-oxopropyl)amine (BOP) / N-nitroso(2-hydroxypropyl) (2-oxopropyl)amine (HPOP)
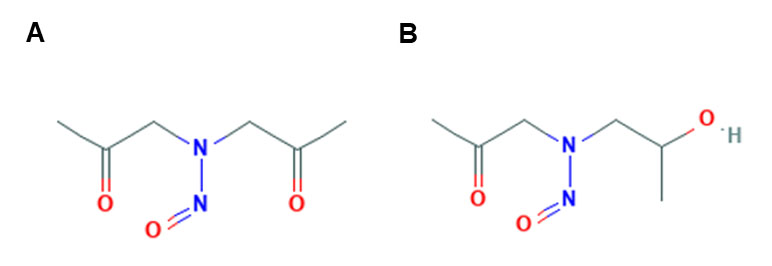
When BOP and HPOP (Figure 1A (3) Figure 1B (4)) are injected subcutaneously (SQ) in Syrian hamsters, they result in the alkylation of DNA and macromolecules in the liver, kidneys, pancreas, and lungs. Thus, these chemicals result in carcinogenesis of not only the pancreas, but also of other organ systems. Two of the most prevalent DNA adducts found were N-methylguanine and O-methylguanine. Based on higher DNA alkylation levels, BOP had more significant carcinogenicity potential compared to HPOP. Some believe BOP is enzymatically activated by reducing BOP to HPOP, but this has been contested (35). Gingell and Pour found that BOP levels and those of its metabolites (HPOP and N-nitroso-bis(2-hydroxypropyl)amine) in the bile, blood, pancreatic juice, and urine were significantly lower after oral (PO) administration compared to intraperitoneal (IP) administration. Given the widespread distribution of the metabolites, it is not surprising that that these chemicals result in different types of cancer throughout the body. Furthermore, because HPOP comprised a large portion of the metabolites, they assumed that it might be a significant contributor of carcinogenesis (24). On the contrary, Lijinsky et al. compared the carcinogenesis of four different nitrosamines, and found that HPOP is not the proximate carcinogenic metabolite and that these compounds induced carcinogenesis through a separate, but unclear mechanism (40).
Figure 1. Chemical structure of BOP (A), and HPOP (B).
Kokkinakis specifically evaluated the difference in BOP and HPOP, comparing Syrian hamsters to rats. They found these compounds to be greater pancreatic carcinogens in Syrian hamsters, while in rats, they had a more significant effect as esophageal and respiratory carcinogens. Furthermore, they found this correlates with DNA alkylation (34). Thus, Syrian hamsters would be a better choice for studying pancreatic cancer compared to rats when using BOP and HPOP to induce carcinogenesis.
Pour administered BOP 20 mg/kg SQ in Syrian hamsters once and then sacrificed hamsters at various time points several weeks after administration. Pour believes centroacinar cells were precursors for pseudoductular formation in the hamster pancreatic cancer model. Electron microscopy and immunohistochemistry demonstrated hypertrophy and hyperplasia of centroacinar cells as early changes; over time, they noted progressive degeneration and loss of acinar cells (53). These pancreatic neoplasms originate from ducts with neoplasia preceded by hyperplasia and dysplasia (51). Takahashi et al. found these changes to occur sequentially: first, necrosis and degeneration of acinar cells; second, hyperplasia of the ductal epithelium; third, development of papillary structures with the rare occurrence of adenocarcinoma at early time points, and a higher predominance of adenocarcinoma at later time points (73). These findings indicate changes in the acinar cells and duct cells as precursor lesions to pancreatic cancer. Fujii et al. found BOP 10 mg/kg SQ administered weekly for four weeks to be ideal for inducing tumors of the lung, liver, nasal cavity, and pancreas with no significant differences related to sex (22). Takiyama et al. used the same regimen as Fujii et al., except they only used male Syrian golden hamsters, with subsequent sacrifice 30 weeks after the initial injection. Takiyama et al. found pseudoductular and ductular adenomas in nearly all hamsters, and ductal carcinoma in situ occurred in 16/18 hamsters (74).
Kokkinakis and Subbarao used female Syrian hamsters and administered a total dose of 210 mg/kg of HPOP via a continuous SQ infusion for nine days, resulting in pancreatic cancer in 62% of these animals after 30 weeks. HPOP had a mitogenic effect in the pancreas with higher DNA adducts, which incite the induction of pancreatic cancer. Overall, they found lower DNA damage in the pancreas than in the liver, kidney, or lung; notably, they found that DNA alkylation alone was not likely to cause ductal tumors (36). Longnecker et al. used 2-week-old Lewis rats and administered HPOP 160 mg/kg IP. They sacrificed the rats at varying time points several months thereafter, and histologically, they found atypical acinar cells, acinar adenomas, and carcinomas. They believe these pancreatic carcinomas derive from acinar cells. Furthermore, they found an equal incidence of liver cell carcinomas and pulmonary adenomas compared to pancreatic carcinomas (43).
Lidjinsky et al. compared carcinogenesis between N-nitroso-2,6-dimethylmorpholine, BOP, N-nitrosobis (2-hydroxypropyl)amine, and HPOP. They gavaged each compound to separate female Syrian golden hamsters, and all four agents resulted in tumors of the pancreas and lungs. BOP was the most potent carcinogen of the four, followed by HPOP; both resulted in hepatocellular and cholangiocellular neoplasms as well (40). Reznik et al. similarly gavaged N-nitroso-2,6-dimethylmorpholine using various dosages, and they found pancreatic tumors in 71% of the hamsters. Here, the carcinogenesis was not dose-dependent. The tumors were either adenomas or adenocarcinomas of ductal origin. However, they also found various tumors of the nasal cavities, upper and lower respiratory tract, liver, gallbladder, renal system, and stomach (59).
Mutations comparable to Humans
In Syrian golden hamsters, BOP results in PDAC with similar histology to human tumors. Additionally, BOP results in genetic mutations and changes in antigens, which correlate clinically with pancreatic cancer in humans. BOP administration in hamsters results in kras mutations, which were identified in hyperplasia (26%), carcinoma in situ (76%), adenocarcinoma (80%), and lymph node (LN) metastases (43%). These mutations were identified through amplified DNA (17). Based on immunohistochemistry, BOP-induced PDAC in hamsters resulted in elevation of the following antigens: 17-1A, CA-125, TAG-72, and DU-PAN-2, all of which are also seen in malignant human pancreatic cancers (74).
Modulators that exacerbate PDAC: Orotic Acid, high fat diet, epidermal growth factor
Sugie et al. found that PDAC can be induced by HPOP, while intraductal papillary mucinous tumors can be induced when HPOP is combined with orotic acid (OA). Female hamsters were administered HPOP 19 mg/kg/day SQ for nine days, and the diet was supplemented with OA for 28 weeks before death or a diet without OA (71). This is an important modulator for those interested in studying intraductal papillary mucinous tumors. Like other models, when BOP is combined with a high-fat diet in hamsters, there is an increased incidence of adenocarcinoma. Specifically, there is an increased incidence of PDAC if a high-fat diet is initiated and continued after BOP administration, while cessation of the high-fat diet before BOP administration showed no difference in cancer incidence than hamsters on a regular diet (14). Notably, pancreatic carcinogenesis was increased 3-4 fold in hamsters on a high-fat diet compared to controls (13). This increase in carcinogenesis associated with high fat diet is logical and has been widely studied. On the other hand, Wouterson et al. showed worse PDAC in hamsters administered BOP in conjunction with dietary fat, while chronic ethanol had no significant effect on carcinogenesis (78). An additional modulator found to exacerbate PDAC was epidermal growth factor (EGF). EGF combined with BOP increased the incidence of PDAC to 75% compared to 44% with BOP alone. Of note, there was no cancer in the EGF without BOP group (18).
Modulators that improve PDAC: Octreotide, tamoxifen, COX-2 inhibitor
Octreotide, a synthetic somatostatin analogue, and tamoxifen, a selective estrogen receptor modulator, have been studied as to their effect on the incidence of PDAC. Octreotide by itself, or in combination with tamoxifen, decreased the incidence of pancreatic cancer and metastatic disease. The combination was not superior to octreotide alone, and there was no difference between the tamoxifen alone compared to the control group (77). Hamsters subjected to BOP have elevated expression of COX-2 through pancreatic carcinogenesis compared to controls. A COX-2 inhibitor was administered to a hamster after a human pancreatic cancer implantation; there was approximately a 5-fold decrease in pancreatic cancer (19). Similarly, a COX-2 inhibitor prevented intraductal papillary carcinoma after hamsters underwent a cholecystoduodenostomy with subsequent BOP administration (10).
B. N-nitrosomethyl(2-oxopropyl)amine (MOP)
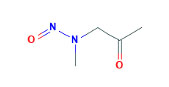
In vitro, MOP (See Figure 2 (7)) has caused significantly more DNA damage when compared to BOP, and pancreatic DNA damage by MOP is mediated through hepatic metabolic activation (37, 67). In vivo, a single MOP 25 mg/kg SQ injection resulted in ductule adenomas or adenocarcinoma in 80% of Syrian golden hamsters. Weekly SQ injections of 3.5 mg/kg or 1.75 mg/kg of MOP for life resulted in pancreatic cancer in 93% and 87% of hamsters, respectively (52). Notably, although MOP is more specific for the pancreas, it is known to cause liver, kidney, vascular, and upper respiratory tract tumors (52).
Figure 2. Chemical structure of MOP.
C. N-nitrosamines 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)
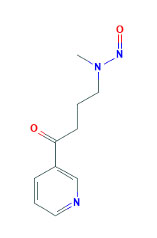
Rivenson et al. demonstrated in F-344 rats that a lifetime supply of NNK (Figure 3 (5)) dissolved in drinking water causes cancer of the exocrine pancreas (acinar adenomas, acinar adenocarcinomas, or ductal adenocarcinomas) along with lung cancer. The mean age of rats at which they saw these pancreatic tumors with the highest concentration of NNK (5.0 ppm) was 90.1+/- 11.8 weeks, but the lung was the principal target organ. They also found tumors of the nasal cavity, liver, testicles, leukemias, lymphomas, carcinoma in situ of the prostate, mammary adenomas, mammary fibromas, mammary adenocarcinomas, thyroid adenocarcinomas, adrenal medullary tumors, skin papillomas, and osteosarcomas. Al-Wadei et al. used a different regimen of NNK 50 mg/kg SQ administration in pregnant Syrian golden hamsters in conjunction with 10% ethanol (ETOH) in drinking water, which resulted in a 65-75% incidence of pancreatitis-associated PDAC (11). 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol NNAL, derived from NNK, resulted in pancreatic tumors in 27% of rats and lung tumors in 87% of rats (60). From genetically engineered mouse models of PDAC, in vitro studies were performed on cell lines derived from pancreatic intraepithelial neoplasia (PanIN), PDAC, and liver metastases. NNK exposure in vitro demonstrated an increase in pCREB expression. In a Ptf1aCreER, LSL-KrasG12D/+ (iPK) mouse model, they found that after administering NNK (10 ppm in drinking water) for 24 weeks demonstrated a significant increase in PanINs compared to controls (70). Osvaldt et al. exposed KrasG12D; Pdx1-Cre (KC) mice to cigarette smoke or clean air for 20 weeks and subsequently performed analyses. KC pancreata exposed to cigarette smoke had elevated levels of PAF1 compared to control controls. Additionally, the same group exposed HPNE and Capan1 cells to cigarette smoke extract (CSE), nicotine and nicotine-derived carcinogens (NNN or NNK), or clean air for 80 days. CSE to cells led to increased expression of ABCg2, Sox9, and PAF1 based on immunohistochemistry and quantitative reverse transcription polymerase chain reaction. Cells exposed to NNN and NNK resulted in increased expression of CHRNA7, FOSL1, and PAF1 as noted on immunoblotting assays (48).
Figure 3. Chemical structure of NNK.
Modulators: Propranolol and ETOH
Propranolol has been found to suppress proliferation and induce apoptosis in a variety of cancers including melanoma, cervical cancer, breast cancer, and liver cancer (25, 47, 75, 81). Al-Wadei et al. found propranolol, a beta-blocker, to have a potent cancer-protective effect by blocking cAMP-dependent intracellular signaling and the release of EGF and vascular endothelial growth factor (VEGF), both of which are known to be implicated in carcinogenesis. Beta-adrenergic receptors are known to result in the proliferation and migration of human PDAC cells in vitro through a cAMP-mediated signaling pathway, potentially activated by NNK, which simultaneously causes the release of epinephrine/norepinephrine in hamsters (11). Ethanol is known to be one of the most common causes of pancreatitis in the world. Thus, it logically follows that ETOH would exacerbate pancreatic ductal adenocarcinoma. The combination of ETOH+NNK demonstrated a statistically significant difference in survival compared to the propranolol group, and the former required euthanasia for anorexia, weight loss, and diarrhea. The ETOH+NNK group also had a significant elevation in VEGF and EGF (11).
D. N-ethyl-N'-nitro-N-nitrosoguanidine (ENNG)
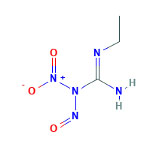
The effects of ENNG (Figure 4 (6)) as a potential carcinogen have been studied in mongrel dogs. These dogs are administered a dose of 595mg of ENNG via a pancreatic intraductal infusion. Sato found that one of four dogs developed ductal adenocarcinoma upon intraductal administration, while the remaining three demonstrated papillary hyperplasia (66). Kamano et al. found pancreatic ductal adenocarcinoma in two of two dogs after a similar intraductal infusion, while in another experiment, they found hyperplasia and “cancerous change of duct epithelial cells” in two of two dogs (30, 31). Overall, this model appears to be poorly studied and the efficacy of the chemical is seemingly low. Further research may yield more meaningful data with evidence of more substantial carcinogenesis induced by this chemical.
Figure 4. Chemical structure of ENNG.
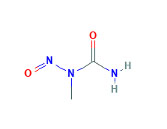
E. N-methyl-N-nitrosourea (MNU)
MNU (Figure 5 (2)) has been long evaluated as a potent model of pancreatic carcinogenesis in hamsters, mice, and guinea pigs. MNU in aqueous solution, used in hamster carcinogenesis studies, spontaneously gives rise to alkylating agents identical to those formed by the activation of BOP and HPOP (35). Furthermore, in vitro studies conducted by Mangold et al. showed that MNU induces a kras mutation at codon 12 or codon 13 (45). In vivo and in vitro studies have demonstrated the carcinogenic properties of MNU. An in vitro model using organ-cultured embryonic rat pancreata with exposure to MNU and 12-O-tetradecanoyl-phorbol-13-acetate resulted in proliferation and hyperplasia of duct-like structures with progression to an abnormal cribriform pattern (50).
Figure 5. Chemical structure of MNU.
In a study by Reddy and Rao, inbred guinea pigs were administered MNU (10 mg/kg) dissolved in sodium citrate buffer via gavage once a week. This resulted in the development of pancreatic adenocarcinoma in 29% of these inbred guinea pigs after 28-44 weeks of administration. Although the pancreas was primarily affected, some guinea pigs also developed gastric adenocarcinoma, colon adenocarcinoma, lymphoma, or hepatoma. Histologically, the exocrine pancreas was abnormal with changes involving acinar ectasia, fibrosis, and increased mitotic activity (57). Previously, Druckrey et al. used outbred guinea pigs and found that the administration of MNU combined with drinking water and urethane resulted in gastric adenocarcinoma and pancreatic adenocarcinoma in approximately 740 days. Specifically, pancreatic adenocarcinoma occurred in 25% of these outbred guinea pigs. Additional findings included sarcomas, leukemia, and metastatic disease (21). Thus, whether using inbred or outbred guinea pigs, MNU has only resulted in the development of pancreatic adenocarcinoma in 25-30% of guinea pigs.
Pancreatic carcinoma has been similarly induced in mice and hamsters. Zimmerman et al. found that intraperitoneal injection of MNU in mice resulted in acinar cell carcinomas in 18% of mice (82). Furukawa et al. showed that after intraperitoneal injections of MNU in Syrian golden hamsters, three histological variants were observed in the pancreas: ductal adenocarcinoma, acinar cell carcinoma, and islet cell carcinoma. Simultaneously, neoplastic lesions were also identified involving the stomach and adrenal gland. They also demonstrated that in hamsters, a single high dose of MNU 50 mg/kg IP had a higher incidence of pancreatic ductal carcinomas than multiple repeated smaller doses (10 mg/kg IP weekly for five weeks) (23).
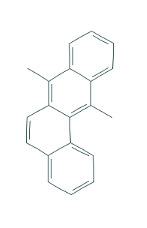
III. 7,12- Dimethylbenz [a]anthracene (DMBA)
DMBA (Figure 6 (1)) is another known carcinogen that has been utilized in various cancer studies. It has been used in mice and rats at a dose of 1mg and 5mg, respectively, with pancreatic implantation of DMBA crystals and subsequent purse-string suturing (12, 16, 29, 49, 76, 80). Alternatively, in mice, Kimura et al. demonstrated that 1mg of DMBA could be dissolved in 0.1% polyoxethylenesorbitan monolaurate and then implanted into the tail of the pancreas with subsequent development of pancreatic cancer (32).
Figure 6. Chemical structure of DMBA.
Pathophysiology
Using Sprague-Dawley rats, Bockman et al. proposed an early generation of tubular complexes, which transform into PDAC. Tubular complexes began to develop as early as two days after implantation; these tubular complexes are generated by acinar cells, duct cells, and some islet cells (16). Jimenez et al. similarly found tubular complexes within one month of implantation. Early changes identified after implantation included edema, inflammatory cell infiltration, fibrosis, hyperplasia, and cell death (16, 29). By one month, there was evidence of PDAC with chronic inflammation marked by a lymphocyte predominance. Immunohistochemistry demonstrated persistent staining for cytokeratin (duct marker) and progressive loss of chymotrypsin (acinar marker) (16). Potential long-term sequela included gastric outlet obstruction, biliary obstruction, and lymph node metastases (29).
Kimura et al. performed DMBA implantation in mice and found tubular complexes within two weeks in 100% of the mice; by one month, 100% of the mice developed metaplastic lesions, by two to three months, 60% developed sarcomatoid carcinomas, and 40% developed dysplastic changes. Furthermore, they believe there was 60% ductal adenocarcinoma by two months, consistent with results in rats (32). Osvaldt et al. had similar findings in mice. Two separate pathologists performed H&E analysis and graded pancreatic lesions as normal, PanIN, or adenocarcinoma. They found PanINs in 67 % and adenocarcinoma in 17% of mice within 30 days. Evaluation at 60 days demonstrated that 35% had PanINs and 38% had PDAC (all pancreata with adenocarcinoma also had PanINs). They also described the phenotype as ductal adenocarcinoma (49). Thus, DMBA is able to result in a large number of cancerous and pre-cancerous lesions after a single application.
The origin of DMBA induced tumors is controversial. Bockman believes that DMBA induced tumors originate from duct cells, progressing from tubular complexes to cancer (15). In contrast, others have described acinar to ductal metaplasia as a prevalent source of carcinogenesis in the DMBA model (16, 29).
Similarities to Human Cancer
Upon immunohistochemical evaluation, DMBA induced pancreatic ductal adenocarcinoma demonstrates a strong expression of cytokeratin, which is similar to human adenocarcinomas (29). Also, after DMBA implantation, immunohistochemistry demonstrates changes in SMAD4, cyclin D1, kras, and p53 expression were noted to be comparable to human pancreatic cancer (32, 49). Additionally, upregulated Notch signaling is frequent in human pancreatic cancer tissues; thus, in mice, continuous activation of Notch signaling may lead to progression from tubular complexes to precancerous lesions to malignant lesions/cancer (32).
Guo et al. induced pancreatic cancer using DMBA in male Sprague-Dawley rats and then performed a gene expression profile using an oligonucleotide microarray with subsequent qRT-PCR and IHC. They noted similarities in upregulation and downregulation of gene expression compared to humans. Upregulated genes included: CXCR7, Clcn3, Kras, cyclinB1, nm23, ATP6v1g2, SMAD4, Acf, Adam6, Cyb5b, FGFR3, Mnd1, Kifc1, Car8, Ptpla, EGFR, Park2, c-Myc, Cadps2, Etv5. Downregulated genes included: Emb, Gabbr1, Zdhhc8, CDKN2a, CXCL10, TP53, Gbp2, Tnfsf9, Dnase1l1, Fgr, Vcan, Lyz, Plod2, Stat5a, Smarca2, Cspg4, Rgs14, Ldb3, Bcl2a1, CCR5 (26).
Modulators that exacerbate PDAC: Cerulein, high fat diet, alcohol, caffeine, nicotine and cigarette smoke
The severity of DMBA-induced pancreatic cancer may be modulated with the addition or utilization of certain substances. For example, Sperti et al. showed the development of adenocarcinoma in 30% of Sprague-Dawley rats after DMBA implantation. However, this number increased to 73% when cerulein (a cholecystokinin analog frequently used in models of pancreatitis) injections were combined with the DMBA implantation. Cerulein is hypothesized to stimulate cell proliferation upon the onset of carcinogenesis (69). In humans, alcohol, nicotine, and a high fat diet are known risk factors for pancreatic cancer. Thus, a high-fat diet led to a significant increase in the incidence of pancreatic ductal adenocarcinoma in male Sprague-Dawley rats after DMBA implantation (80).
In mice, alcohol, caffeine, nicotine, and cigarette smoke also are important disease modulators. Wendt et al. showed that after DMBA implantation, administration of alcohol combined with caffeine (23.8%) or administration of alcohol alone (52.9%) led to significantly more adenocarcinoma compared to administration of water only (16.6%) or administration of caffeine only (15%) group Bersch et al. performed DMBA implantation in mice after exposure to nicotine or cigarette smoke for 16 days, continued the nicotine or cigarette smoke, and euthanized thirty days after surgery. PDAC occurred at a higher frequency in the DMBA + nicotine group (51.9%) compared to DMBA + cigarette smoke group (13.3%) and DMBA only group (16.7%) (12).
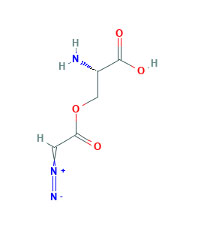
IV. Azaserine (O-diazoacetyl-L-serine)
Another chemical model of carcinogenesis is azaserine (Figure 7 (8)), which has been studied in both hamsters and rats. Azaserine is a bacterial mutagen and pancreatic carcinogen in Wistar rats, and it is believed to be a monofunctional alkylating agent, which subsequently alkylates DNA. Azaserine 5 mg/kg IP has been administered to Wistar/Lewis Rats once or twice a week for six months with subsequent sacrifice in 1-2 years. This resulted in pancreatic carcinomas in 47% of cases, renal tumors in 27% of cases, and hepatomas in 5%. It causes irreparable damage preferentially to pancreatic tissue DNA (41). Doses of 10 mg/kg IP and 30 mg/kg IP resulted in comparable DNA damage, but lower doses (3 mg/kg) resulted in less consistent DNA damage (41, 42).
Figure 7. Chemical structure of Azaserine.
Longnecker and Curphey found that within the 1st year of administering azaserine 5 mg/kg IP, there were areas of the atypical exocrine pancreas marked by hyperplastic foci, encapsulated adenomas, and hyperplastic nodules. Furthermore, >25% had pancreatic adenocarcinoma with metastasis to lymph nodes, liver, and lung. Other tumor sites included the breast, neck, liver, pituitary gland, subcutaneous tissues, and the rectum (42). Histological changes of the exocrine pancreas become evident in as little as two months, and adenomas become apparent six months after treatment (41).
Roebuck et al. administered a single 30 mg/kg IP injection in male Lewis rats and sacrificed the rats at 6, 9, 12, 15, and 18 months thereafter. After nine months, 30% of rats demonstrated carcinoma in situ, while 18 months after administration, there was a 100% incidence of pancreatic cancer (58% carcinoma in situ and 42% carcinoma) (61).
Longnecker et al. used a different regimen in Wistar and Wistar/Lewis rats, administering 5-10 mg/kg/week IP or PO for 52 weeks with subsequent sacrifice immediately after that or after an additional year. They found that individual tumors measured as large as 6.5 cm in diameter, equally affecting the head, body, and tail. Histologically, they found acinar cell carcinomas of either a pure or mixed phenotype: duct-like carcinoma, undifferentiated carcinoma, cystic carcinoma, and microadenocarcinoma. They also noted breast cancers in female rats, ear duct tumors, and hepatomas (44).
Modulators that exacerbate PDAC: Bombesin, cerulein, cholecystokinin, diet, and testosterone
Some modulators, when combined with azaserine, increase acinar cell nodules or pre-neoplastic lesions. These include bombesin, cerulein, and cholecystokinin (20, 38, 46). Other studies have shown modulation of the effect of Azaserine via dietary supplementation of various compounds. Azaserine combined with a high essential fatty acids diet led to an increase in foci of acidophilic foci compared to basophilic foci and pre-neoplastic lesions, which are thought to progress to adenomas and adenocarcinomas (64). Roebuck et al. also found that the effect of azaserine is enhanced by unsaturated fat, raw soy protein isolate, and corn oil supplementation (63). Roebuck et al. demonstrated calorie-restricted diets, high protein diets, and diets high in unsaturated fat increased pancreatic neoplasms (65). Similarly, a high-fat diet leads to worse pancreatic ductal adenocarcinoma. While ethanol led to increased multifocal disease, it did not exacerbate or increase the incidence of carcinogenesis in rats (78).
Testosterone levels also appear to play a role in the degree of carcinogenesis. Lhost et al. identified the effect of steroid hormones on azaserine-induced pancreatic cancer, and they concluded that males have a higher incidence of these tumors. Furthermore, testosterone may result in worse carcinogenesis, while estrogen may play a protective role (39). These findings may explain the sex-based differences in these cancers. Similarly, Longnecker et al. found more pancreatic and renal carcinomas in male rats than female rats (79).
Modulators that improve PDAC: Retinoids, acetylsalicylic acid, and retinyl acetate
When combined with azaserine, certain substances were found to have a protective effect against pancreatic cancer. Some inhibitors of pancreatic cancer include retinoid supplementation (vitamin A derivative), acetylsalicylic acid (non-steroidal anti-inflammatory drug [NSAID]), and retinyl acetate (vitamin A derivative) (44, 62, 79). Vitamin A derivatives were evaluated because they have been found to decrease the incidence of certain cancers, or potentially delay the progression in chemical induced models of cancer (44). Lorglumide, a cholecystokinin receptor antagonist, inhibited the exacerbating effects of cholecystokinin combined with azaserine but not of bombesin combined with azaserine (46).
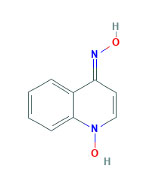
V. 4-Hydroxyaminoquinoline-1-oxide
4-hydroxyaminoquinoline-1-oxide (Figure 8 (9)) is another chemical-induced carcinogenesis model studied in f344 rats, ICR mice, Hartley Albino guinea pigs, and Syrian golden hamsters. Qin et al. evaluated organs three hours after administering 10 mg/kg intravenous (IV) in all four species, and rats showed the highest DNA binding in the pancreas and kidney. Furthermore, after a dose of 6 mg/kg IV, histologic changes became evident in rats while mice required 24 mg/kg IV. Guinea pigs and hamsters did not show any significant pancreatic histologic changes (54). Thus, rats would be a more cost effective model compared to mice.
Figure 8. Chemical structure of 4-hydroxyaminoquinioline-1-oxide.
Rats seem especially sensitive to this model. Shinozuka et al. demonstrated that a single IV injection in rats resulted in multiple atypical acinar cell nodules upon evaluation at 6, 9, and 12 months after injection (68). Others have evaluated rat pancreata after a shorter duration. Kitagawa et al. administered a single IV dose of 14 mg/kg in male rats and evaluated them after 24, 48, 72, and 168 hours. Their findings included large vacuoles, decreased zymogen granules, RER alterations, acinar cell changes, increasing amounts of necrosis, nuclear disorganization, interstitial space-filling with inflammatory cells, and duct-like cells. They noted that there was damage up to 48 hours with some degree of repair and
regeneration between 72 and 168 hours (33).
Rao et al. performed a single IV injection in male Wistar rats at a dose of 6 mg/kg, and they found atypical acinar cell foci with increased mitotic activity and irregular plasma membranes in 100% of rats (56). Others have evaluated the genetic alterations that predispose these rats to the development of cancer. Imazawa et al., for example, found that after administration of a single 20 mg/kg IV dose to male rats, based on immunohistochemistry, p53 expression and apoptosis were noted to be significantly increased 4-6 hours after, and the rate peaked at 24 hours. Furthermore, they found proliferation, evaluated through ki67, peaked at 72 hours (28).
When applied to mice, a single IV dose of 24 mg/kg led to 100% of mice developing atypical acinar cell foci with an associated increase in mitotic activity. These findings are similar to lesions seen in rat pancreata after induction (55). Reddy et al. found a single IV dose of 22.5 mg/kg resulted in necrosis of the exocrine pancreas and elevated serum amylase levels in an inbred strain of guinea pigs after 48 hours. Maximal necrosis occurred at 48 hours, and there was a subsequent regenerative phase with complete restoration noted by 84 hours (58).
Modulators of PDAC: Testosterone
Testosterone also modulates the carcinogenesis in this model. A single IV dose of 9 mg/kg resulted in a higher rate of pancreatic adenomas in male rats compared to female rats. Furthermore, castration in males decreased the risk in male rats, while administering testosterone in females increased the risk (27).
VI. Discussion
In this review article we have thoroughly examined the literature and briefly summarized our review of various chemical induced animal models of pancreatic cancer, including nitrosamines, DMBA, azaserine, and 4-hydroxyaminoquinoline-1-oxide. There is a vast literature that has been published regarding these compounds, but we attempted to summarize some of the most relevant research experiments pertinent to both clinical and basic science research.
There are multiple factors to consider when deciding to proceed with a chemical induced model of pancreatic cancer. Certain chemicals to induce pancreatic cancer are far more widely studied than the others. For example, 4-hydroxyaminoquinoline-1-oxide appeared to result in pancreatic cancer, however, the actual experiments showed successful carcinogenesis in very small numbers, which raises questions about the efficacy of this model.
Other considerations to be taken into account are off target effects of the chemicals. Metastatic pancreatic cancer is a separate disease entity compared to primary tumors of other origin. Some of these chemicals induce primary tumors of organs other than the pancreas, which depending on the experimental design or necessity, this may or may not be desired. This may be valuable when studying cancerous states in which multiple organ systems are involved. Furthermore, for those studying the progression of pancreatic cancer, it is logical to study metastatic disease, however, if the chemical results in offsite carcinogenesis, understanding the pathophysiology becomes muddied and unclear.
Another important consideration is familiarly with the animal model species and the route of administration of the chemical. Special training may be required for certain species depending on individual IACUC protocols, requiring learning about new animal species, their handling, and handling of these potential carcinogens. Furthermore, certain chemicals require surgical implantation while others can be administered via oral, SQ, IV, or IP routes. This is also an important consideration and should be used to determine the ease and comfort of proceeding with a certain model.
Additionally, another two important matters to be taken into account are the desired timeframe and the cost. Depending on the route of administration, some of these models are much more expeditious and result in severe cancers quickly. Others require multiple doses of the chemical over a prolonged time to result in a pancreatic cancer after a longer duration. Both are potentially fruitful depending on the intended usage whether it be for shorter duration or a longer duration. Also, cost is also an important factor to contemplate. Smaller animals are usually cheaper than larger animals. However, there are additional housing costs and costs of food that should also be taken into account. In addition, some of these chemicals are cheaper than others, depending on the chosen regimen and planned frequency.
Arguably, the most important consideration is the purpose of the planned experiments. These models are powerful because they allow researchers to investigate novel therapies. Pancreatic cancer is a devastating malignancy, and new therapies are necessary. These therapies may be used primarily or as adjuvant therapy to attempt to completely eliminate the cancer. Other important arms to potentially investigate include a prophylactic arm, whereby a therapy is applied in a preventative fashion before the onset of a cancer, and a palliative arm, where a therapy is used to treat advanced cancers to either increase survival or decrease the pancreatic cancer symptoms.
We have learned a great deal about pancreatic cancer. However, these models may help provide new insight into the pathophysiology and disease process of pancreatic cancer. Different chemicals will result in different premalignant lesions/precursor lesions, cancers originating from the exocrine pancreas, and cancers originating from the endocrine pancreas. This will potentially allow for the identification of implicated mutations and their subsequent outcomes in various types of pancreatic cancer. Subsequently, better understanding of different types of pancreatic cancer will allow for the generation of better therapies able to treat and cure cancer.
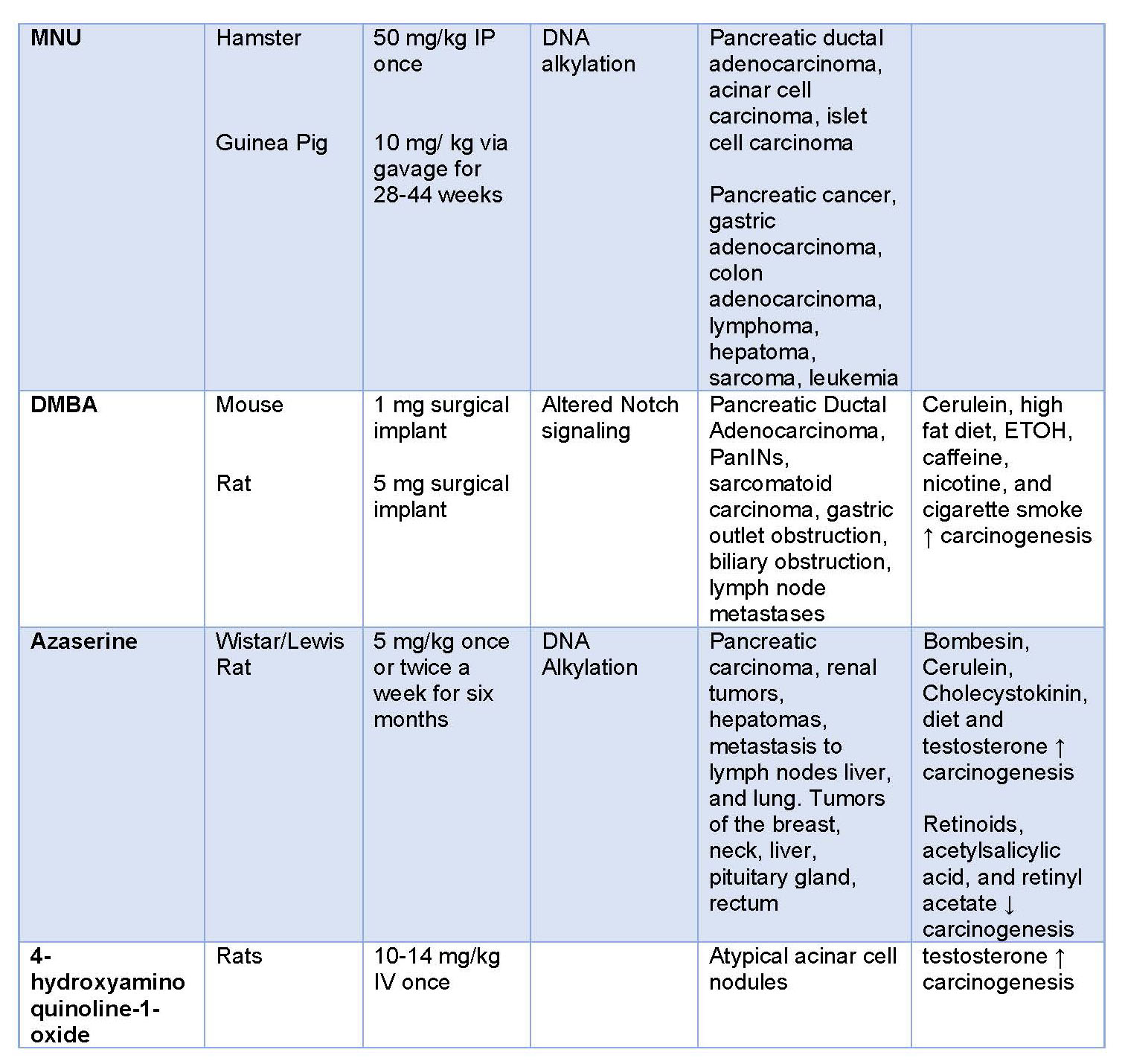
In summary, there are numerous chemical induced models that can be utilized for various purposes. There are multiple considerations one should take into account. The advantages and disadvantages of the models are summarized in Table 1.
Table 1. Advantages and disadvantages of carcinogen models of PDAC.
VII. References
- PubChem Compound Summary for CID 6001, 7,12-Dimethylbenz[a]anthracene. PubChem: National Center for Biotechnology Information, 2021.
- PubChem Compound Summary for CID 12699, N-Nitroso-N-methylurea. PubChem: National Center for Biotechnology Information, 2021.
- PubChem Compound Summary for CID 43371, N-Nitrosobis(2-oxopropyl)amine. PubChem: National Center for Biotechnology Information, 2021.
- PubChem Compound Summary for CID 43677, N-Nitroso(2-hydroxypropyl)(2-oxopropyl)amine. PubChem: National Center for Biotechnology Information, 2021.
- PubChem Compound Summary for CID 47289, 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone. PubChem: National Center for Biotechnology Information, 2021.
- PubChem Compound Summary for CID 93349, N'-Ethyl-N-nitro-N-nitrosoguanidine. PubChem: National Center for Biotechnology Information, 2021.
- PubChem Compound Summary for CID 104910, N-Nitrosomethyl(2-oxopropyl)amine. PubChem: National Center for Biotechnology Information, 2021.
- PubChem Compound Summary for CID 460129, azaserine. PubChem: National Center for Biotechnology Information, 2021.
- PubChem Compound Summary for CID 5360001 4-(Hydroxyamino)quinoline 1-oxide. PubChem: National Center for Biotechnology Information, 2021.
- Adachi T, Tajima Y, Kuroki T, Mishima T, Kitasato A, Tsuneoka N, and Kanematsu T. Chemopreventive effects of a selective cyclooxygenase-2 inhibitor (etodolac) on chemically induced intraductal papillary carcinoma of the pancreas in hamsters. Carcinogenesis 29: 830-833, 2008. PMID: 18296437.
- Al-Wadei HA, Al-Wadei MH, and Schuller HM. Prevention of pancreatic cancer by the beta-blocker propranolol. Anticancer Drugs 20: 477-482, 2009. PMID: 19387337.
- Bersch VP, Osvaldt AB, Edelweiss MI, Schumacher Rde C, Wendt LR, Abreu LP, Blom CB, Abreu GP, Costa L, Piccinini P, and Rohde L. Effect of nicotine and cigarette smoke on an experimental model of intraepithelial lesions and pancreatic adenocarcinoma induced by 7,12-dimethylbenzanthracene in mice. Pancreas 38: 65-70, 2009. PMID: 18824948.
- Birt DF, Julius AD, White LT, and Pour PM. Enhancement of pancreatic carcinogenesis in hamsters fed a high-fat diet ad libitum and at a controlled calorie intake. Cancer Res 49: 5848-5851, 1989. PMID: 2790796.
- Birt DF, Salmasi S, and Pour PM. Enhancement of experimental pancreatic cancer in Syrian golden hamsters by dietary fat. J Natl Cancer Inst 67: 1327-1332, 1981. PMID: 6273636.
- Bockman DE. Cells of origin of pancreatic cancer: experimental animal tumors related to human pancreas. Cancer 47: 1528-1534, 1981. PMID: 6268274.
- Bockman DE, Guo J, Büchler P, Müller MW, Bergmann F, and Friess H. Origin and development of the precursor lesions in experimental pancreatic cancer in rats. Lab Invest 83: 853-859, 2003. PMID: 12808120.
- Cerny WL, Mangold KA, and Scarpelli DG. K-ras mutation is an early event in pancreatic duct carcinogenesis in the Syrian golden hamster. Cancer Res 52: 4507-4513, 1992. PMID: 1643642.
- Chester JF, Gaissert HA, Ross JS, and Malt RA. Pancreatic cancer in the Syrian hamster induced by N-nitrosobis(2-oxopropyl)-amine: cocarcinogenic effect of epidermal growth factor. Cancer Res 46: 2954-2957, 1986. PMID: 3009003.
- Crowell PL, Schmidt CM, Yip-Schneider MT, Savage JJ, Hertzler DA, 2nd, and Cummings WO. Cyclooxygenase-2 expression in hamster and human pancreatic neoplasia. Neoplasia 8: 437-445, 2006. PMID: 16820089.
- Douglas BR, Woutersen RA, Jansen JB, de Jong AJ, Rovati LC, and Lamers CB. Influence of cholecystokinin antagonist on the effects of cholecystokinin and bombesin on azaserine-induced lesions in rat pancreas. Gastroenterology 96: 462-469, 1989. PMID: 2910761.
- Druckrey H, Ivankovic S, Bucheler J, Preussmann R, and Thomas C. [Production of stomach and pancreatic cancer in the guinea pig using methylnitroso-urea and methylnitroso-urethane]. Z Krebsforsch 71: 167-182, 1968. PMID: 4233463.
- Fujii K, Hayakawa T, and Kikuchi M. Tumor induction in mice administered neonatally with bis(2-oxopropyl)nitrosamine. Tohoku J Exp Med 174: 361-368, 1994. PMID: 7732518.
- Furukawa F, Sato H, Imaida K, Toyoda K, Imazawa T, Takahashi M, and Hayashi Y. Induction of pancreatic tumors in male Syrian golden hamsters by intraperitoneal N-methyl-N-nitrosourea injection. Pancreas 7: 153-158, 1992. PMID: 1553365.
- Gingell R, and Pour P. Metabolism of the pancreatic carcinogen N-nitroso-bis(2-oxopropyl)amine after oral and intraperitoneal administration to Syrian golden hamsters. J Natl Cancer Inst 60: 911-913, 1978. PMID: 633398.
- Gong L, Lei Y, Tan X, Dong Y, Luo Z, Zhang D, and Han S. Propranolol selectively inhibits cervical cancer cell growth by suppressing the cGMP/PKG pathway. Biomed Pharmacother 111: 1243-1248, 2019. PMID: 30841438.
- Guo JC, Li J, Yang YC, Zhou L, Zhang TP, and Zhao YP. Oligonucleotide microarray identifies genes differentially expressed during tumorigenesis of DMBA-induced pancreatic cancer in rats. PLoS One 8: e82910, 2013. PMID: 24376604.
- Hayashi Y, and Katayama H. Promoting effect of testosterone propionate on experimental exocrine pancreatic tumors by 4-hydroxyaminoquinoline 1-oxide in rats. Toxicol Lett 9: 349-354, 1981. PMID: 6800070.
- Imazawa T, Nishikawa A, Toyoda K, Furukawa F, Mitsui M, and Hirose M. Sequential alteration of apoptosis, p53 expression, and cell proliferation in the rat pancreas treated with 4-hydroxyaminoquinoline 1-oxide. Toxicol Pathol 31: 625-631, 2003. PMID: 14585730.
- Jimenez RE, Z'Graggen K, Hartwig W, Graeme-Cook F, Warshaw AL, and Fernandez-del Castillo C. Immunohistochemical characterization of pancreatic tumors induced by dimethylbenzanthracene in rats. Am J Pathol 154: 1223-1229, 1999. PMID: 10233860.
- Kamano T, Azuma N, Katami A, Tamura J, Sakakibara N, Matsumoto M, Mizumoto K, Kitazawa S, and Konishi Y. Preliminary observation on pancreatic duct adenocarcinoma induced by intraductal administration of N-ethyl-N'-nitro-N-nitrosoguanidine in dogs. Jpn J Cancer Res 79: 1-4, 1988. PMID: 3128497.
- Kamano T, Tamura J, Uchida T, Kanno T, Sakakibara N, Tsutsumi M, Maruyama H, and Konishi Y. Studies by pancreatography of ductal changes induced by administration of pancreatic carcinogen in two dogs. Jpn J Clin Oncol 21: 282-286, 1991. PMID: 1942557.
- Kimura K, Satoh K, Kanno A, Hamada S, Hirota M, Endoh M, Masamune A, and Shimosegawa T. Activation of Notch signaling in tumorigenesis of experimental pancreatic cancer induced by dimethylbenzanthracene in mice. Cancer Sci 98: 155-162, 2007. PMID: 17297654.
- Kitagawa T, Okamura K, Sohma M, Namiki M, and Ono K. Ultrastructural changes in pancreatic acinar cells of rats after administration of 4-hydroxyaminoquinoline-1-oxide. Histol Histopathol 1: 369-375, 1986. PMID: 2980132.
- Kokkinakis DM. Differences in DNA-guanine alkylation between male Sprague-Dawley rats and Syrian hamsters following exposure to a single dose of pancreatic nitrosamine carcinogens. Chem Res Toxicol 3: 150-156, 1990. PMID: 2130943.
- Kokkinakis DM, and Scarpelli DG. DNA alkylation in the hamster induced by two pancreatic carcinogens. Cancer Res 49: 3184-3189, 1989. PMID: 2720674.
- Kokkinakis DM, and Subbarao V. The significance of DNA damage, its repair and cell proliferation during carcinogen treatment in the initiation of pancreatic cancer in the hamster model. Cancer Res 53: 2790-2795, 1993. PMID: 8504421.
- Lawson T, and Nagel D. The production and repair of DNA damage by N-nitrosobis(2-oxopropyl)amine and azaserine in hamster and rat pancreas acinar and duct cells. Carcinogenesis 9: 1007-1010, 1988. PMID: 3370746.
- Lhoste EF, and Longnecker DS. Effect of bombesin and caerulein on early stages of carcinogenesis induced by azaserine in the rat pancreas. Cancer Res 47: 3273-3277, 1987. PMID: 3581068.
- Lhoste EF, Roebuck BD, Brinck-Johnsen T, and Longnecker DS. Effect of castration and hormone replacement on azaserine-induced pancreatic carcinogenesis in male and female Fischer rats. Carcinogenesis 8: 699-703, 1987. PMID: 3581428.
- Lijinsky W, Saavedra JE, Knutsen GL, and Kovatch RM. Comparison of the carcinogenic effectiveness of N-nitrosobis(2-hydroxypropyl)amine, N-nitrosobis(2-oxopropyl)amine, N-nitroso(2-hydroxypropyl)(2-oxopropyl)amine, and N-nitroso-2,6-dimethylmorpholine in Syrian hamsters. J Natl Cancer Inst 72: 685-688, 1984. PMID: 6583451.
- Lilja HS, Hyde E, Longnecker DS, and Yager JD, Jr. DNA damage and repair in rat tissues following administration of azaserine. Cancer Res 37: 3925-3931, 1977. PMID: 908032.
- Longnecker DS, and Curphey TJ. Adenocarcinoma of the pancreas in azaserine-treated rats. Cancer Res 35: 2249-2258, 1975. PMID: 1097106.
- Longnecker DS, Roebuck BD, Kuhlmann ET, and Curphey TJ. Induction of pancreatic carcinomas in rats with N-nitroso(2-hydroxypropyl)(2-oxopropyl)amine: histopathology. J Natl Cancer Inst 74: 209-217, 1985. PMID: 3871492.
- Longnecker DS, Roebuck BD, Yager JD, Jr., Lilja HS, and Siegmund B. Pancreatic carcinoma in azaserine-treated rats: induction, classification and dietary modulation of incidence. Cancer 47: 1562-1572, 1981. PMID: 6974040.
- Mangold KA, Hubchak S, Mangino MM, Laconi S, and Scarpelli DG. In vitro carcinogenesis of hamster pancreatic duct cells: cellular and molecular alterations. Carcinogenesis 15: 1979-1984, 1994. PMID: 7923593.
- Meijers M, Appel MJ, van Garderen-Hoetmer A, Lamers CB, Rovati LC, Jansen JB, and Woutersen RA. Effects of cholecystokinin and bombesin on development of azaserine-induced pancreatic tumours in rats: modulation by the cholecystokinin receptor antagonist lorglumide. Carcinogenesis 13: 1525-1528, 1992. PMID: 1394835.
- Montoya A, Varela-Ramirez A, Dickerson E, Pasquier E, Torabi A, Aguilera R, Nahleh Z, and Bryan B. The beta adrenergic receptor antagonist propranolol alters mitogenic and apoptotic signaling in late stage breast cancer. Biomed J 42: 155-165, 2019. PMID: 31466709.
- Nimmakayala RK, Seshacharyulu P, Lakshmanan I, Rachagani S, Chugh S, Karmakar S, Rauth S, Vengoji R, Atri P, Talmon GA, Lele SM, Smith LM, Thapa I, Bastola D, Ouellette MM, Batra SK, and Ponnusamy MP. Cigarette Smoke Induces Stem Cell Features of Pancreatic Cancer Cells via PAF1. Gastroenterology 155: 892-908.e896, 2018. PMID: 29864419.
- Osvaldt AB, Wendt LR, Bersch VP, Backes AN, de Cássia ASR, Edelweiss MI, and Rohde L. Pancreatic intraepithelial neoplasia and ductal adenocarcinoma induced by DMBA in mice. Surgery 140: 803-809, 2006. PMID: 17084724.
- Parsa I, Marsh WH, and Sutton AL. An in vitro model of pancreas carcinogenesis: two-stage MNU--TPA effect. Am J Pathol 98: 649-662, 1980. PMID: 7361849.
- Pour P, Althoff J, and Takahashi M. Early lesions of pancreatic ductal carcinoma in the hamster model. Am J Pathol 88: 291-308, 1977. PMID: 195472.
- Pour P, Gingell R, Langenbach R, Nagel D, Grandjean C, Lawson T, and Salmasi S. Carcinogenicity of N-nitrosomethyl(2-oxopropyl)amine in Syrian hamsters. Cancer Res 40: 3585-3590, 1980. PMID: 7438045.
- Pour PM. Mechanism of pseudoductular (tubular) formation during pancreatic carcinogenesis in the hamster model. An electron-microscopic and immunohistochemical study. Am J Pathol 130: 335-344, 1988. PMID: 3277441.
- Qin XS, Nakatsuru Y, Tada M, and Ishikawa T. Species and organ differences in DNA adduct formation and repair after treatment with 4-hydroxyaminoquinoline 1-oxide. Jpn J Cancer Res 81: 613-619, 1990. PMID: 2119365.
- Rao MS, Subbarao V, and Scarpelli DG. Atypical acinar cell lesions of the pancreas in mice induced by 4-hydroxyaminoquinoline-1-oxide. Int J Pancreatol 2: 1-10, 1987. PMID: 2890694.
- Rao MS, Upton MP, Subbarao V, and Scarpelli DG. Two populations of cells with differing proliferative capacities in atypical acinar cell foci induced by 4-hydroxyaminoquinoline-1-oxide in the rat pancreas. Lab Invest 46: 527-534, 1982. PMID: 6804715.
- Reddy JK, and Rao MS. Pancreatic adenocarcinoma in inbred guinea pigs induced by n-methyl-N-nitrosourea. Cancer Res 35: 2269-2277, 1975. PMID: 1149036.
- Reddy JK, Rao MS, Svoboda DJ, and Prasad JD. Pancreatic necrosis and regeneration induced by 4-hydroxyaminoquinoline-1-oxide in the guinea pig. Lab Invest 32: 98-104, 1975. PMID: 1113507.
- Reznik G, Mohr U, and Lijinsky W. Carcinogenic effect of N-nitroso-2,6-dimethylmorpholine in Syrian golden hamsters. J Natl Cancer Inst 60: 371-378, 1978. PMID: 202716.
- Rivenson A, Hoffmann D, Prokopczyk B, Amin S, and Hecht SS. Induction of lung and exocrine pancreas tumors in F344 rats by tobacco-specific and Areca-derived N-nitrosamines. Cancer Res 48: 6912-6917, 1988. PMID: 3180100.
- Roebuck BD, Baumgartner KJ, and Longnecker DS. Growth of pancreatic foci and development of pancreatic cancer with a single dose of azaserine in the rat. Carcinogenesis 8: 1831-1835, 1987. PMID: 3499996.
- Roebuck BD, Baumgartner KJ, Thron CD, and Longnecker DS. Inhibition by retinoids of the growth of azaserine-induced foci in the rat pancreas. J Natl Cancer Inst 73: 233-236, 1984. PMID: 6610790.
- Roebuck BD, Kaplita PV, Edwards BR, and Praissman M. Effects of dietary fats and soybean protein on azaserine-induced pancreatic carcinogenesis and plasma cholecystokinin in the rat. Cancer Res 47: 1333-1338, 1987. PMID: 3815341.
- Roebuck BD, Longnecker DS, Baumgartner KJ, and Thron CD. Carcinogen-induced lesions in the rat pancreas: effects of varying levels of essential fatty acid. Cancer Res 45: 5252-5256, 1985. PMID: 3876880.
- Roebuck BD, Yager JD, Jr., and Longnecker DS. Dietary modulation of azaserine-induced pancreatic carcinogenesis in the rat. Cancer Res 41: 888-893, 1981. PMID: 7459874.
- Sato T. [Experimental study of pancreatic duct adenocarcinoma in dog]. Nihon Geka Gakkai Zasshi 92: 1486-1492, 1991. PMID: 1961186.
- Schaeffer BK, Wiebkin P, Longnecker DS, Coon CI, and Curphey TJ. DNA damage produced by N-nitrosomethyl(2-oxopropyl)amine (MOP) in hamster and rat pancreas: a role for the liver. Carcinogenesis 5: 565-570, 1984. PMID: 6609783.
- Shinozuka H, Popp JA, and Konishi Y. Ultrastructures of atypical acinar cell nodules in rat pancreas induced by 4-hydroxyaminoquinoline-1-oxide. Lab Invest 34: 501-509, 1976. PMID: 818451.
- Sperti C, Militello C, Rovati L, Behboo R, Khajeturian E, Perasole A, Alaggio R, and Pedrazzoli S. Effect of cholecystokinin analogue caerulein and cholecystokinin antagonist lorglumide on pancreatic carcinogenesis in the rat. J Surg Oncol 57: 11-16, 1994. PMID: 8065144.
- Srinivasan S, Totiger T, Shi C, Castellanos J, Lamichhane P, Dosch AR, Messaggio F, Kashikar N, Honnenahally K, Ban Y, Merchant NB, VanSaun M, and Nagathihalli NS. Tobacco Carcinogen-Induced Production of GM-CSF Activates CREB to Promote Pancreatic Cancer. Cancer Res 78: 6146-6158, 2018. PMID: 30232221.
- Sugio K, Gazdar AF, Albores-Saavedra J, and Kokkinakis DM. High yields of K-ras mutations in intraductal papillary mucinous tumors and invasive adenocarcinomas induced by N-nitroso(2-hydroxypropyl)(2-oxopropyl)amine in the pancreas of female Syrian hamsters. Carcinogenesis 17: 303-309, 1996. PMID: 8625455.
- Surveillance E, and End Results (SEER) Program (www.ser.cancer.gov) SEER*Stat Database: Incidence - SEER Research Data. Cancer Stat Facts: Pancreatic Cancer. 2020.
- Takahashi M, Pour P, Althoff J, and Donnelly T. Sequential alteration of the pancreas during carcinogenesis in Syrian hamsters by N-nitrosobis(2-oxopropyl)amine. Cancer Res 37: 4602-4607, 1977. PMID: 922742.
- Takiyama Y, Egami H, and Pour PM. Expression of human tumor-associated antigens in pancreatic cancer induced in Syrian hamsters. Am J Pathol 136: 707-715, 1990. PMID: 2316628.
- Wang F, Liu H, Wang F, Xu R, Wang P, Tang F, Zhang X, Zhu Z, Lv H, and Han T. Propranolol suppresses the proliferation and induces the apoptosis of liver cancer cells. Mol Med Rep 17: 5213-5221, 2018. PMID: 29393410.
- Wendt LR, Osvaldt AB, Bersch VP, Schumacher Rde C, Edelweiss MI, and Rohde L. Pancreatic intraepithelial neoplasia and ductal adenocarcinoma induced by DMBA in mice: effects of alcohol and caffeine. Acta Cir Bras 22: 202-209, 2007. PMID: 17546293.
- Wenger FA, Kilian M, Mautsch I, Jacobi CA, Schimke I, Saul GJ, Guski H, and Müller JM. Influence of octreotide and tamoxifen on tumor growth and liver metastasis in N-nitrosobis(2-oxopropyl)amine-induced pancreatic cancer in Syrian hamsters. Horm Res 54: 74-77, 2000. PMID: 11251370.
- Woutersen RA, van Garderen-Hoetmer A, Bax J, and Scherer E. Modulation of dietary fat-promoted pancreatic carcinogenesis in rats and hamsters by chronic ethanol ingestion. Carcinogenesis 10: 453-459, 1989. PMID: 2924393.
- Yıldız H, Oztas H, Yıldız D, Koc A, and Kalipci E. Inhibitory effects of acetylsalicylic acid on exocrine pancreatic carcinogenesis. Biotech Histochem 88: 132-137, 2013. PMID: 23331184.
- Z'Graggen K, Warshaw AL, Werner J, Graeme-Cook F, Jimenez RE, and Fernández-Del Castillo C. Promoting effect of a high-fat/high-protein diet in DMBA-induced ductal pancreatic cancer in rats. Ann Surg 233: 688-695, 2001. PMID: 11323507.
- Zhou C, Chen X, Zeng W, Peng C, Huang G, Li X, Ouyang Z, Luo Y, Xu X, Xu B, Wang W, He R, Zhang X, Zhang L, Liu J, Knepper TC, He Y, and McLeod HL. Propranolol induced G0/G1/S phase arrest and apoptosis in melanoma cells via AKT/MAPK pathway. Oncotarget 7: 68314-68327, 2016. PMID: 27582542.
- Zimmerman JA, Trombetta LD, Carter TH, and Weisbroth SH. Pancreatic carcinoma induced by N-methyl-N'-nitrosourea in aged mice. Gerontology 28: 114-120, 1982. PMID: 7084676.
