
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2016.8
| Attachment | Size |
|---|---|
| 775.88 KB |
1. Introduction
Dysregulation of multiple signaling pathways in the pancreas is proposed to be involved the initiation of pancreatitis. Genetic manipulation in experimental animals has been used to understand the critical roles of these pathways during the pathogenesis of pancreatitis. In general, the genetic approaches that specifically target these specific molecules are superior to pharmacological methods. Additionally, genetic modification of the initiator genes observed in human pancreatitis can help create clinically relevant animal models. These models will be valuable for studying the mechanisms of pancreatitis and testing preclinical therapeutic and preventive interventions. In this review, we will focus on the impact of trypsin, NF-kB, ER stress, Ras, autophagy, Cox-2 and several others on pancreatitis. We will also discuss efforts to create animal models for human hereditary pancreatitis, cystic fibrosis, and autoimmune pancreatitis.
2. Animal Models Targeting Trypsinogen Activation
Trypsin and Pancreatitis
The exocrine pancreas synthesizes digestive enzymes to facilitate digestion. More than a century ago, Chiari (21) proposed that acute pancreatitis (AP) was an autodigestive disease. During the past two decades, researchers discovered that trypsinogen is prematurely activated in various animal models of pancreatitis and human pancreatitis (13, 39, 68, 72, 137). Because trypsin is capable of degrading proteins and initiating other zymogen activation cascades, premature activation of trypsinogen in pancreatic acinar cells is considered a key initiator of this disease. This notion is strongly supported by the observation that gain-of-function cationic trypsinogen (PRSS1) mutations and loss-of-function mutations of the potent pancreatic protease inhibitor Kazal type 1 (SPINK1) are associated with hereditary pancreatitis (HP) (8, 14, 110, 129, 132, 133, 139, 140). The mechanisms of trypsinogen activation have been extensively studied (45, 100, 109, 127). However, the roles of intracellular trypsin have only recently been directly examined. In transgenic mouse models, expression of active trypsin or mutant PRSS1 caused pancreatitis, and the expression of the trypsin inhibitor SPINK1 ameliorated experimental pancreatitis (8, 37, 87, 106). Genetic deletion of trypsinogen 7, the most prominent trypsinogen in mice, blunted trypsinogen activation and caused a 50% reduction in acinar necrosis following caerulein challenges (25).
Models with Direct Trypsinogen Activation
Because pharmacological inhibitors have nonspecific effects, and the stimuli used to induce pancreatitis activate multiple signaling pathways in addition to activating trypsinogen (48), the direct effects of intracellular trypsin activity cannot be examined using these approaches. However, a recent study reported the development of a mutant trypsinogen that could be activated intracellularly by the endogenous protease PACE (paired basic amino acid cleaving enzyme) (61) (Fig. 1A). This new construct (PACE-trypsinogen) allowed for the first time direct examination of the effects of intracellular trypsin on pancreatic acinar cells. In in vitro studies, this PACE-trypsinogen was expressed by means of adenoviral vector in the secretory pathway and was activated within acinar cells. Expression of PACE-trypsinogen induced the apoptosis of pancreatic acinar cells. Cell death was blocked by the trypsin inhibitor pefabloc, but it was not completely blocked by the pancaspase inhibitor benzyloxycarbonyl-VAD, indicating that caspase-independent pathways were also involved. However, intracellular trypsin had no significant effect on the activity of the proinflammatory transcription factor Nuclear Factor kappa B (NF-κB). In contrast, extracellular trypsin caused cell damage and dramatically increased NF-κB activity. These data support that the effects of active trypsin on pancreatic acinar cells are determined by its localization (61). For in vivo studies, a new mouse model was developed with this construct. These mice were engineered to conditionally express an endogenously activated trypsinogen within pancreatic acinar cells after cre mediated recombination (37) (Fig. 1B). The acinar specificity was achieved by crossing these mice with mice harboring pancreatic elastase I promoter-driven CreERT, a tamoxifen-inducible cre (63) (Fig. 2A). Commonly used pancreatic specific cre mice were summarized in Fig. 2B. Interestingly, initiation of AP was observed with high (homozygous) but not low (heterozygous) expression levels of PACE-trypsinogen. Rapid caspase-3 activation and apoptosis with delayed necrosis was observed, and lost acinar cells were replaced with abundant fatty tissue and limited fibrosis. These findings indicated that intra-acinar activation of trypsinogen is sufficient to initiate AP. This novel model will provide a powerful tool for improving our understanding of the basic mechanisms that occur during the initiation of pancreatitis (37).

Fig 1. Expression of a trypsinogen that can be activated intracellularly. A. WT trypsinogen is normally activated in duodenum by enteropeptidase (top). A mutant trypsinogen was developed by an insertion of amino acid sequence RTKR immediate before the active trypsin. RTKR sequence can be recolonized and cleave by PACE . B. In a conditional transgenic mouse line, PACE-trypsinogen expression was blocked by a loxp-GFP-stop-loxP cassette until Cre medicated recombination (37, 61).
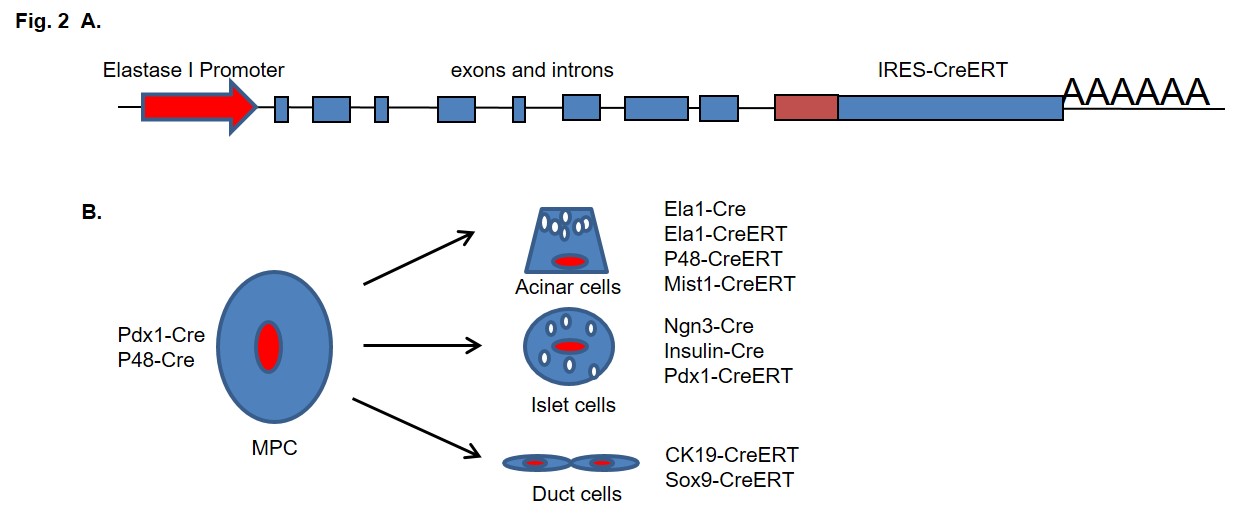
Fig 2. Commonly used cell specific Cre mouse lines for pancreatic research. A. For inducible pancreatic acinar specific gene targeting, a tamoxifen-inducible Cre (CreERT) was introduced to a Bacterial Artificial Chromosome containing the pancreatic elastase I gene. The full length promoter of elastase I gave high efficient and specific CreERT expression in acinar cells. B. pdx1 and p48 (Ptf1a) are transcription factors expressed early in pancreatic multipotent progenitor cells (MPC). Cre driven by these promoters will be active in acinar, ductal and endocrine cells. Pancreatic elastase I promoter is pancreatic acinar cells specific. Although P48 and Mist1 are activated early in MPC, in adults they are expressed only in acinar cells. Therefore, using tamoxifen inducible CreERT system in adult, these gene promoters will only induce recombination in pancreatic acinar cells. Similarly, Pdx1 promoter is active only in endocrine cells in adult pancreas. Ngn3 drives the differentiation of endocrine islet. Cre expression driven by this gene promoter will be all pancreatic endocrine lineages. Insulin promoter is specific to beta cells. For pancreatic duct cells, CK19 and Sox9 promoters can be used. However, these genes are also expressed in many other organs.
A Model with SPINK Inactivation
SPINK1 (Serine Protease Inhibitor Kazal-type 1), also known as pancreatic secretory trypsin inhibitor (PSTI), was originally identified as a trypsin inhibitor in 1948 by Kazal et al. (65) SPINK1 is synthesized in the acinar cells of the pancreas and binds to trypsin to prevent further activation of pancreatic enzymes. Thus, a lack of SPINK1 may result in the premature conversion of trypsinogen into active trypsin in acinar cells, leading to autodigestion of the exocrine pancreas by activated proteases. The detailed functions and roles of SPINK1in pancreatic diseases have been reviewed previously (80, 95). Since Witt et al first showed that mutations in the SPINK1 gene were associated with chronic pancreatitis in 2000, (139) there have been many reports on the association between mutations in SPINK1 genes and patients with pancreatitis (10, 70, 97).
In mice, SPINK3 (Serine Protease Inhibitor Kazal type 3) is the homolog of human SPINK1. Upregulation of SPINK 3 provides a protective mechanism in caerulein-induced pancreatitis (90), and knockdown of SPINK3 (Spink3−/− mice) enhanced trypsin activity in pancreatic acinar cells (93). The pancreata of Spink3−/− mice develop usually up to 15.5 days after fertilization; however, autophagic degeneration of acinar cells, but not ductal or islet cells, begins after 16.5 days. Rapid onset of cell death occurs in the pancreas and duodenum within a few days after birth and results in death by 14.5 days after birth. There is limited inflammatory cell infiltration and no signs of apoptosis. At 7.5 days after birth, residual ductlike cells in the tubular complexes strongly express pancreatic duodenal homeodomain-containing protein 1, a marker of pancreatic stem cells, without any sign of acinar cell regeneration. These findings indicate that the progressive disappearance of acinar cells in Spink3−/− mice was due to autophagic cell death and impaired regeneration, suggesting that SPINK3 maintains the integrity and regeneration of acinar cells (94). Although SPINK3 is also expressed in the kidney, lung, and a small proportion of cells in the gastrointestinal tract and liver (111), pancreas-specific transgenic expression of SPINK rescues SPINK3-/- mice and restores a normal pancreatic phenotypes, supporting the critical role of SPINK3 in pancreatic homeostasis (87, 105).
Hereditary Pancreatitis-Related Models
Hereditary pancreatitis (HP) is a rare form of pancreatitis (118). The first family with HP was described by Comfort and Steinberg in 1952 (23). Using genetic linkage studies, the HP locus was independently narrowed to the long arm of chromosome 7 by Le Bodic, Whitcomb, and Pandya in 1996 (71, 98, 134). Shortly after, Whitcomb et al (133) identified an Arg-His substitution at residue 117 of the cationic trypsinogen gene (PRSS1) in HP through mutational analysis. It was originally named R117H based on the chymotrypsin numbering system and was renamed R122H following the recommendations for gene mutation nomenclature (7, 20). Subsequently, a number of gain-of-function PRSS1 mutations were identified. The most common ones were R122H, A16V, and N29I (88). Clinically, HP is characterized by recurrent AP with an unusual early onset of the disease (5–23 years) and the development of chronic pancreatitis. Importantly, the cumulative risk of pancreatic cancer after the onset of symptoms is 44% at 70 years after the onset of the disease in HP patients. The calculated standardized incidence ratio of pancreatic cancer from the European registry of hereditary pancreatitis and pancreatic cancer (EUROPAC) cohort after correction for age, history of smoking, nationality, and surgical intervention was 67 (130).
Efforts have been made by several groups to generate an animal model mimicking the mutations harbored in HP patients with limited success. In one model, human PRSS1 mutant R122H transgenic mice were generated under using a 213-bp fragment of the rat elastase promoter/enhancer. PRSS1 was expressed in low amounts in zymogen granules. No spontaneous development of pancreatitis was observed. However, serum pancreatic lipase levels or activity was higher in these animals after induction of pancreatitis compared to controls. Repeated caerulein insults resulted in a slightly more severe pancreatitis. This rather small difference in comparison to controls could be caused by the low expression of the transgene in the mouse pancreas (114).
Another transgenic mouse was generated in which the expression of the mouse PRSS1 mutant R122H (R122H_mPRSS1) was expressed using a rat elastase promoter (8). Acinar cell damage was detectable by 7 weeks of age with increasing inflammatory infiltrates at 12 weeks. Fibrosis was evident in 24-week-old animals. The ultimate penetrance of this phenotype reached 40% after 1 year of age. The inflammatory phenotype varied in both extent and severity among the animals. It is surprising that the rapid response of the transgenic animals to caerulein injection was indistinguishable from that of the WT. In contrast, 1 week after injection the inflammatory phenotype in the WT animals largely resolved, while the R122H_mPRSS1 transgenic animals displayed extensive collagen deposition in the periacinar and interlobular areas, indicating a chronic inflammatory response. This is the first genetic model of chronic pancreatitis in which a genetic alteration resulted in a predicted phenotype. Unfortunately, there have been no follow-up studies published since 2006. Therefore, this model might be “lost” during breeding, probably due to unstable gene expression from the small elastase promoter.
In another recent study, Athwal et al developed a transgenic mouse model system using WT human PRSS1 and two HP-associated mutants (R122H and N29I) using a rat elastase promoter (9). The transgenic animals revealed pathological changes similar to that of chronic pancreatitis particularly in aging (> 9 months) animals. These changes occurred spontaneously in up to 10% of the animals that expressed the transgenes. When a supra-physiological dose (50 μg/kg) of caerulein was administered to the WT and PRSS1 transgenic strains, no differences in pancreatic response were observed. However, when these animals were treated with a lower dose of caerulein (20 μg/kg), the transgenic animals from each of the three transgenic strains displayed more severe pancreatitis than WT animals. The transgenic expression of PRSS1 promoted apoptotic rather than necrotic cell death. It is intriguing that no differences in phenotype were observed among these transgenic strains and that the supra-physiological dose of caerulein did not cause more severe pancreatitis in the transgenic mice compared to the WT mice.
3. Animal Models Induced by Modulating NF-κB Activity
Introduction of Nuclear Factor Kappa B (NF-κB)
The NF-κB transcription factor family is expressed in almost all cell types and tissues, and specific NF-κB binding sites are present in the promoters/enhancers of a large number of genes. These genes regulate inflammation, immunity, cell proliferation, differentiation, and survival (28). NF-κB consists of five proteins, p65 (RelA), RelB, c-Rel, p105/p50 (NF-κB1), and p100/52 (NF-κB2) that associate with each other to form distinct transcriptionally active homo- and hetero- dimeric complexes. Among these, p65, RelB, and c-Rel contain c-terminal transactivation domains. p50 and p52 are generated by processing of the precursor molecules p105 and p100, respectively. The p50/65 heterodimer represents the most abundant form of Rel dimers and is expressed in almost all cell types. However, not all combinations of Rel dimers are transcriptionally active: DNA-bound p50 and p52 homo- and hetero-dimers have been found to repress κB-dependent transcription. In most unstimulated cells, NF-κB dimers are retained in an inactive form in the cytosol through their interaction with IκB proteins. Degradation of these inhibitors upon their phosphorylation by the IκB kinase (IKK) complex leads to nuclear translocation of NF-κB and induction of the transcription of target genes (53, 54, 86, 115).
The canonical IKKs, IKKα and IKKβ, form a complex with the regulatory adaptor protein NF-κB essential modulator (NEMO; also known as IKKγ). IKKα and IKKβ are Ser/Thr kinases that phosphorylate the NF-κB inhibitor IκB proteins, resulting in their poly-ubiquitination and subsequent proteasomal degradation. This allows NF-κB to translocate to the nucleus and bind to specific DNA elements. Despite extensive sequence similarity, IKKα and IKKβ have largely distinct functions due to their different substrate specificities and modes of regulation. IKKβ (and IKKγ) are essential for rapid NF-κB activation by proinflammatory signaling cascades, such as those triggered by tumor necrosis factor alpha (TNFα) or lipopolysaccharide (LPS). In contrast, IKKα participates in the activation of a specific form of NF-κB in response to a subset of TNF family members and may also serve to attenuate IKKβ-driven NF-κB activation (46). Based on sequence similarities with IKKα and IKKβ, two IKK-related kinases, TANK binding kinase 1 (TBK1) and IKKɛ (also known as IKK-inducible or IKK-i), were discovered. IKKɛ and TBK1 are known as non-canonical IKKs. These protein kinases are important for the activation of interferon response factors 3 and 7. NF-κB is activated in most inflammatory diseases including animal models of AP (41, 108). Moreover, IKKs are also involved in kinase and NF-κB-independent activities (46, 92, 116, 128).
Activation of IKK2 (IKKB) Induced Pancreatitis
To specifically address the roles of NF-κB in pancreatitis, the active form of NF-κB subunit can be expressed in the pancreas. IKK contains a canonical MAP kinase kinase activation loop motif in which phosphorylation of both serine residues is necessary for activation. Mutation of both the serine residues to glutamate residues mimics the effect of phospho-serine and generates highly active kinase activity (26, 83). An early attempt to conditionally overexpress the constitutively active mutant of IKK2 in the pancreas used a tetracycline-inducible system. To achieve transgene expression in the pancreas, tetO-IKK2-EE (EE denotes serine to glutamate mutations) animals were crossed with CMV-reverse tetracycline-responsive transactivator (rtTA) mice (66). In these double transgenic animals, doxycycline treatment induced expression of IKK2-EE in pancreatic acinar cells, resulting in moderate activation of the IKK complex. IKK2 expression in the pancreas had a mosaic pattern, and the activation level of the NF-κB cascade induced by IKK2 was considerably lower compared with that observed after supramaximal caerulein stimulation, but it still led to the formation of the leucocyte infiltrates observed after 4 weeks of doxycycline stimulation. The infiltrates were mainly composed of B lymphocytes and macrophages. However, only minor damage to pancreatic tissue was observed, indicating that a moderate level of activation is not sufficient to induce pancreatic damage in mice (4).
The influence of expression and thus activation level of NF-κB on the development of AP was confirmed by a second study from the same group of investigators published a few months later. In the new transgene system, transgenic mice expressing the rtTA gene under the control of a rat elastase promoter were generated to mediate acinar cell-specific expression of IKK2 alleles. Expression of dominant-negative IKK2 ameliorated caerulein-induced pancreatitis but did not affect trypsinogen activation. Expression of constitutively active IKK2 was sufficient to induce AP, including increased edema, cellular infiltrates, necrosis, serum lipase levels, and pancreatic fibrosis (11). These phenotypes were likely caused by higher expression levels from the tandem (tetO)7 Promoter (50).
Activation of the IKK2/NF-κB signaling pathway causing AP is further confirmed by transgenic mice that encode the NF-κB p65 subunit or constitutively active IKK2 in the pancreatic acinar cells (57). Transgenic expression of p65 led to compensatory expression of the inhibitory subunit IΚB-α. Therefore, there is no increased NF-κB activity or clear phenotype. However, p65 transgenic mice had higher levels of NF-κB activity in acinar cells and greater levels of inflammation in pancreatic tissue upon caerulein challenge. In contrast, pancreas-specific expression of active IKK2 directly increased the activity of NF-κB in acinar cells and induced pancreatitis. Prolonged activity of IKK2 resulted in the activation of stellate cells, loss of acinar cells, and initiation of fibrosis, which are the characteristics of chronic pancreatitis. Co-expression of IKK2 and p65 further increased the expression levels of inflammatory mediators and the severity of pancreatitis (57). In addition, this pathway also orchestrated oncogenic Ras-induced inflammation and tumorigenesis, which will be further discussed below (24).
Overall, these findings indicate that NF-κB activity increased the severity of pancreatic inflammation, and strategies to inactivate NF-κB may be used to treat patients with acute or chronic pancreatitis. However, selective truncation of the p65 gene (Fig 3), which leads to the reduction the NF-κB activity, in pancreatic exocrine cells surprisingly led to both severe injury of the acinar cells and systemic adverse events (6). This is because the expression and induction of the protective pancreas-specific acute phase protein pancreatitis-associated protein 1 (PAP1) depends on RelA/p65. Lentiviral gene transfer of PAP1 cDNA reduced the severity of pancreatitis in mice with selective truncation of RelA/p65. Opposing functions of RelA/p65 on AP in different cell types have been reported. For example, RelA/p65 activation in myeloid cells promotes the pathogenesis of CP but protects against chronic inflammation in acinar cells (126).

Fig 3. An example of tissue specific gene deletion. Two identical loxp sites flank several exons of p65 gene. Cre mediated recombination will remove the sequences between these two sites, causing gene truncation or deletion.
The collected data argue that NF-κB functions more generally as a central regulator of stress responses and regulates both inflammation and cell survival. Coupling stress responsiveness and anti-apoptotic pathways through the use of a common transcription factor may result in increased cell survival following stress insults (92).
Functional Roles of IKK1 (IKKα)
IκB kinase α (IKKα) is a subunit of the IKK complex, which functions together with IKKβ and IKKγ/NEMO. Although it is involved in the regulation of NK-κB activity by phosphorylation of IκB proteins, which triggers their degradation, IKKα is not required for degradation of IκB by proinflammatory stimuli. IKKα is more important in alternative NF-κB signaling in which RelB:p52 dimers are activated (46). Global loss of IKKα perturbs multiple morphogenetic events, including limb and skeletal patterning and the proliferation and differentiation of epidermal keratinocytes (46, 55, 107). Pancreatic-specific IKKα ablation (using PDX-CRE) results in acinar cell vacuolization and death, fibrosis, and inflammation, resembling chronic pancreatitis in humans. These studies support that IKKα plays a critical role in maintaining pancreatic acinar cell homeostasis (74, 75). The role of IKKα in maintaining pancreatic homeostasis is independent of NF-κB or its protein kinase activity reflected in inactive IKK1 knock-in mutant mice, which exhibit no pancreatic abnormalities. In contrast, the loss of IKKα in acinar cells diminished autophagic protein degradation and caused the accumulation of p62 aggregates and ER stress. Pancreatic specific p62 ablation ameliorated pancreatitis in IKKα deficient mice (74).
IκB-α Mutation/Deletion and Pancreatitis
One of the key target genes induced by NF-κB is its inhibitor IκBα, which in turn inhibits NF-κB activity and thus establishes a feedback regulation mechanism for controlling NF-κB activity (120). Therefore, IκB-α deficiency results in a sustained NF-κB response and severe widespread dermatitis in mice (67). Although they appear normal at birth, IκB-α-/- mice exhibit severe runting, skin defects, and extensive granulopoiesis postnatally and typically die after 8 days (12).
In a mouse model where the two well-defined κB enhancer elements and four additional κB-like sites in the IκBα promoter were altered and therefore defective in response to NF-κB activation-induced negative feedback, the mice became sick, had elevated serum cytokine levels, and died at 13 to 15 months of age. These mice were found to be hypersensitive to LPS-induced proinflammatory cytokine production and lethality. Pancreas, liver, and lung tissues derived from these mice at 3 months of age showed extensive perivascular lymphocytic infiltration. Immunohistochemical analysis revealed a phenotype resembling Sjögren’s syndrome, an autoimmune disorder (99).
Pancreas-specific ablation of IκB-α also led to increased basal NF-κB activity with small increases in cytokine and chemokine levels. In stark contrast, the basal increase of NF-κB did not cause any overt phenotype in the pancreas but caerulein- and L-arginine–induced pancreatitis was ameliorated in IΚB-α-/- mice (89). The amelioration of pancreatitis was lost in the mice when p65 was also ablated, supporting that the protective effects of IκB deletion were through p65. These results were consistent with this group’s previous study reporting that pancreas-specific RelA/p65 truncation increases the susceptibility of acini to inflammation-associated cell death following caerulein-induced pancreatitis (6). They concluded that acinar cell NF-κB activation exerts a protective role in AP.
In summary, NF-κB regulates both cell survival and inflammation. The extent, mode of NF-κB manipulation, and cellular context can have profoundly different consequences. The paradox that activation of IKK2 or p65 leads to both aggravation and amelioration of pancreatitis reflects the complicated roles of NF-κB signaling. The complexities of NF-κB system, in both pro- and anti-inflammatory effects, have been discussed in an editorial (43).
Ras Signaling and Pancreatitis
Ras proteins function as binary molecular switches that, when turned on by occupancy with GTP, interact with downstream signaling molecules to activate a wide variety of intracellular signaling networks regulating proliferation, differentiation, apoptosis, and cell migration (56). Ras mutations lead to increased Ras activity and are observed in ~30% of all cancers. In particular, K-Ras mutations are found in nearly every pancreatic cancer, which is the fourth leading cause of cancer-related death in USA (117). The mutant Ras is oncogenic as has been shown in many in vivo and in vitro studies (119). However, its role in the development of inflammation and fibrosis has not been well appreciated until recently (24, 64, 76).
It is well known that activating Ras mutations can induce either proliferation at low activity or senescence at high-activity levels. In mice bearing targeted pancreas-specific activating mutations at the native K-Ras locus, which increases Ras activity at lower level, pancreatic histology was normal at early stages. Only a minor subset of cells displayed hyperproliferation and tumorigenesis when the levels of Ras activity were significantly elevated (2). Evidence for the existence of a threshold of Ras pathway activity in pancreatic pathology comes from studies expressing higher levels of mutant K-Ras using a transgenic approach (Fig. 4A) (64). Although the expression of endogenous levels of mutant K-Ras results in minimal change in the pancreas, higher levels of expression generate Ras pathway activity that mimics what is observed in pancreatic cancer cells. In this model, increased Ras activity in pancreatic acinar cells caused rapid and abundant development of inflammatory cell infiltration, fibrosis, acinar cell loss, and atrophy. This model confirms that elevated levels of Ras activity are sufficient to cause chronic pancreatitis-like changes. These data suggest that the activity level of the Ras pathway, rather than the presence of mutations, is biologically important.
Transgenic overexpression of mutant K-Ras provides proof-of-concept that high Ras activity will cause an inflammatory response with drastic fibrosis. However, under physiological conditions, human or mice with knock-in mutant Ras only express endogenous levels of mutant K-Ras. While the expression of oncogenic Ras at physiological levels using a K-Ras knock-in model generally does not directly cause pathological outcomes (Fig. 4B) (52), the presence of mutations predispose Ras activity to be further significantly elevated by various etiologic stimulations (24).

Fig 4. Different expression levels in transgene and knock-in models. A. Mutant KRas was expressed at high level from a strong CMV chimeric promoter (pCAG) after Cre recombination. B. A mutation was knocked into the endogenous KRas exon (*). Upon Cre recombination, the mutant KRas was expressed from its native promoter at physiological level.
The stimuli that only cause transient up-regulation of Ras activity in WT animals induced enhanced and prolonged stimulation in animals bearing oncogenic K-Ras. Importantly, this increased Ras signaling led to the development of similar chronic inflammatory changes as observed in transgenic K-Ras mice with high Ras activity. Further study demonstrated that inflammatory or physiologic stimuli triggered an NF-κB–mediated positive feedback mechanism involving Cox-2 that amplified Ras activity to pathological levels. Because a large portion of the adult human population possess Ras mutations, disruption of this positive feedback loop may be an important strategy for cancer prevention (24). It may be puzzling why stimulation is needed for 'constitutively active' mutant Ras to increase the activity and cause pathology, but it has been demonstrated that oncogenic K-Ras is not constitutively active, as has been believed, but can be readily activated by upstream stimulants leading to prolonged strong Ras activity (56). These data indicate that in addition to targeting K-Ras downstream effectors, interventions to reduce K-Ras activation may have important cancer-preventive value, especially in patients with oncogenic Ras mutations.
4. ER Stress and Pancreatitis
Pancreatic acinar cells are specialized for the production of a large amount of digestive enzymes. The enzyme proteins are produced in the endoplasmic reticulum (ER), a multifunctional organelle responsible for the synthesis and folding of proteins in the secretory pathway. When misfolded proteins in the ER exceed the capacity of ER chaperones (eg. GRP78), the unfolded protein response (UPR) will be activated. In mammalian cells, the UPR is transduced by three ER-localized transmembrane protein sensors: activating transcription factor 6α (ATF6α), inositol-requiring kinase 1α (IRE1α), and PKR-like ER kinase (PERK). ATF6α leads to increased transcription of ER chaperones, including GRP78 (BiP) and protein foldases. Activation of IRE1 induces the splicing and activation of XBP1 to its active transcription factor form (sXBP1), which also regulates the expression of various chaperones, foldases, and other protective molecules. PERK activation leads to phosphorylation of eIF-2α and results in decreased protein translation. At a certain level, the UPR mechanisms decrease misfolded proteins and alleviate stress by shutting down translation and upregulating protective molecules. However, prolonged and severe UPRs can lead to apoptosis by upregulating the transcription factor C/EBP-homologous protein (CHOP) and the IRE1-activated JNK pathway. Severe ER stress also impairs cellular homeostasis through ER calcium leakage, mitochondrial damage, oxidative stress, energy depletion, and activation of caspases. ER stress triggered by protein misfolding represents a potential disease mechanism for pancreatitis (1, 32, 38, 69, 78, 109). Genetic deletion of GRP78 leads to peri-implantation lethality (79). Grp78 heterozygosity regulates ER chaperone balance, in dietary- and genetic background-dependent manners, and improved ER protein folding may be protective against pancreatitis (141). Pancreata that have a genetic deletion of CHOP are histologically normal, and the role of CHOP in pancreatitis remains controversial (124, 131).
XBP1 and Pancreatitis
Pancreatic acinar-specific disruption of Xbp1 in adult pancreatic acinar cells with Mist1-CreERT led to the activation of the UPR, extensive apoptosis followed by a rapid phase of recovery that included the expansion of the centroacinar cell compartment, formation of tubular complexes that contained Hes1- and Sox9-expressing cells, and regeneration of acinar cells that expressed Mist1 from the residual, surviving Xbp1+ cell population. This study suggests that XBP1 is required for the homeostasis of adult acinar cells in mice (51). In ethanol-fed Xbp1+/− mice, ER stress was associated with disorganized and dilated ER, loss of zymogen granules, accumulation of autophagic vacuoles, and increased acinar cell death (77).
PERK and Pancreatitis
The exocrine and endocrine pancreas developed normally in Perk−/− mice. Postnatally, ER distention and activation of the ER stress transducer IRE1α accompanied increased cell death and led to progressive diabetes mellitus and exocrine pancreatic insufficiency (49). In another study with pancreatic specific PERK ablation (Ela-CRE), there was no evidence for perturbations in ER-stress but acinar cells succumbed to a non-apoptotic form of cell death, oncosis, which is associated with a pronounced inflammatory response and induction of pancreatitis stress response genes (58). In the same study, the effects of ATF4 on the pancreas were evaluated in ATF4 deficient mice because the translation of the ATF4 transcription factor is positively regulated by PERK activation. In these mice, the exocrine pancreata of neonatal (P4) mice were severely underdeveloped and the number of acini and the acinar cell size were greatly reduced. The pancreatic acini were dispersed and often were not in close proximity to neighboring acini, resulting in an expanded extracellular space. Moreover, ATF4 deficient acinar cells appeared smaller with substantially less zymogen granule content, which correlated with an increase in the diameter of the centroacinar duct. In adults the centroacinar duct was greatly expanded resulting in a tubular appearance of the exocrine pancreas with numerous adipocytes (58).
5. Cystic Fibrosis
Cystic fibrosis (CF) is caused by mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR). The CFTR protein is highly expressed in pancreatic duct epithelia. CFTR is an ion channel that conducts chloride and thiocyanate ions across epithelial cell membranes. Mutations in the CFTR gene lead to dysregulation of epithelial fluid transport in the lung, pancreas and other organs. In the CF pancreas, a high protein concentration due to a low flow of secretions causes precipitation in the duct leading obstruction and damage. These changes in the CF pancreas begin in utero. Eventually, this process results in the obstruction of ducts by mucus, the destruction of acini, severe inflammation, generalized fibrosis, and fat replacement (138).
Mouse Models of CF
Since the discovery of CFTR gene in 1989 (102), many genetic mouse models have been developed to study the pathophysiology of CF and test experimental therapies prior to clinical trials (31, 136). All of the CFTR-mutant mice developed prominent intestinal disease, similar to what is seen in human CF, and most mice die from intestinal obstruction during the first month of life. Although the lung and pancreas are the organs severely affected in human CF, mouse models with CFTR mutations lack obvious lung and pancreas histology except in one study (136). In one study, Cftr-/- mice were weaned to a liquid diet to minimize bowel obstruction and optimize long-term viability. Under these conditions, the intercalated, intralobular and interlobular ducts and acinar lumina of the exocrine pancreas of the Cftr-/- animals were dilated and filled with inspissated material. There was also mild inflammation and acinar cell drop out. Quantitative measurements of the pancreas showed significant acinar atrophy and increased acinar volume in comparison with age-matched WT littermates (33). Cftr-/- mice were also more sensitive to caerulein-induced severe pancreatitis than WT mice (29, 30).
When human CFTR (hCFTR) was expressed in Cftr-/- mice under the control of the rat intestinal fatty acid-binding protein gene promoter, the mice survived and showed functional correction of ileal goblet cell and crypt cell hyperplasia and cyclic adenosine monophosphate-stimulated chloride secretion. These results support the concept that transfer of the hCFTR gene may be a useful strategy for correcting physiologic defects in patients with CF (143).
Pig Models of CF
Because pigs share many anatomical and physiological features with humans, pigs with CFTR gene disruptions or mutations (DeltaF508) were generated by adeno-associated virus (rAAV)-mediated gene targeting (103). Newborn pigs lacking CFTR exhibited defective chloride transport and developed meconium ileus, exocrine pancreatic destruction, and focal biliary cirrhosis, replicating the abnormalities seen in newborn humans with CF. The Cftr-/- porcine pancreas was smaller than WT controls. Microscopic examination revealed small, degenerative lobules with increased loose adipose and myxomatous tissue, as well as scattered to moderate cellular inflammation. Residual acini had diminished amounts of eosinophilic zymogen granules. Ducts were variably dilated and obstructed by eosinophilic material plus neutrophils and macrophages mixed with cellular debris. Pancreatic endocrine tissue was spared. These changes are similar to those in human CF (59, 104). Cftr DF508/DF508 pancreata had reduced parenchyma compared to those in Cftr +/+ mice, but the destruction was slightly less severe than in Cftr-/- because this mutant has residual CFTR activity (96). The pig model may provide opportunities to address persistent questions about CF pathogenesis and accelerate the discovery of strategies for prevention and treatment.
Ferret Model of CF
The domestic ferret is also an alternative species to be used for modeling human CF because ferrets and humans share similar airway cytoarchitecture and CFTR gene expression patterns. A CFTR gene-deficient model in the domestic ferret has been developed using rAAV -mediated gene targeting of exon 10 in fibroblasts and nuclear transfer cloning (123). Neonatal CFTR-knockout ferrets demonstrated many of the characteristics of human CF disease. The pancreata newborn Cftr-/- ferrets were normal at the gross level, but histologic lesions were evident with acinar lumen and duct dilation in all animals. Overall, the level of histopathology in the newborn Cftr-/- ferret pancreas appears to be quite similar to that seen in CF infants and significantly less severe than the extensive destruction observed in the exocrine pancreas of newborn CF pigs (122). Further evaluation of older CF animals indicated that 85% of these CF animals had significant loss of the exocrine pancreas with associated fibrosis, ductal proliferation, and plugging of intralobular ducts. Interestingly, some of the CF animals had fewer histological changes with only focal loss of exocrine parenchyma and cystic dilation of ducts. These findings demonstrate that the ferret model retains the variability in pancreatic phenotypes also seen in CF patients, where 10% to 15% of CF patients retain partial or complete exocrine pancreas function. Modifier genes are the most likely explanation for the pancreatic sufficiency (121).
6. Inflammatory Factors/Growth Factors/Cytokines and Pancreatitis
Transforming Growth Factor-Beta (TGF-β)
TGF-β 1 proteins are central regulators of pancreatic cell function and play key roles in pancreatic development and diseases (101). In transgenic mice that express TGF-β 1 in the pancreatic islet cells by use of a human insulin promoter, fibroblast proliferation and abnormal deposition of macrophages and neutrophils in the extracellular matrix were observed from birth on, which replaced almost the entire exocrine pancreas. TGF-β 1 inhibited proliferation of acinar cells and resulted in small islet cell clusters. These findings suggest that TGF-β 1 might be a mediator of diseases associated with extracellular matrix deposition, such as chronic pancreatitis (73).
In a mouse model in which TGF-β signaling was inactivated in mouse pancreas by overexpressing a dominant-negative mutant form of the TGF-β type II receptor in the pancreas under the control of the pS2/TFF1 promoter, the mice showed marked increases in MHC class II molecules and matrix metalloproteinase expression in pancreatic acinar cells. These mice also showed increased susceptibility to caerulein-induced pancreatitis. Therefore, TGF-β signaling seems to be essential either for maintaining normal immune homeostasis and suppressing autoimmunity or for preserving the integrity of pancreatic acinar cells (47). Remarkably, in another study from the same group, attenuated caerulein-induced pancreatic fibrosis was reported in these mice (142).
The dominant-negative mutant type II TGF-β receptor (DNR), the extracellular and transmembrane domains of TβRII, blocks signaling of all three TGF-β isoforms. In transgenic mice expression of DNR with a mouse MT1 promoter, the pancreas showed severe abnormalities, including ductular transformation, neo-angiogenesis, inter- and intralobular fibrosis and adipose replacement of acini in the exocrine pancreas (16). These mice exhibited reduced pancreatitis in response to caerulein compared to wild-type control mice, indicating that a functional TGF-beta signaling pathway may be required for caerulein to induce AP (135).
In mice with conditional knockout of the TGFβ type II receptor by an S100A4/fibroblast-specific protein 1 (FSP1) Cre, which is expressed in dendritic cells (DC) and fibroblasts, autoimmune pancreatitis (AIP) spontaneously developed by 6 weeks of age. Adoptive transfer of bone marrow-derived DC from Tgfbr2 KO mice into 2-week-old syngeneic WT mice resulted in reproduction of pancreatitis within 6 weeks. In contrast, adoptive transfer of Tgfbr2 KO DC to adult mice failed to induce pancreatitis, suggesting a developmental event in AIP pathogenesis. This model illustrates the role of TGF β for maintaining myeloid DC immune tolerance (15).
Cyclooxygenase-2 (COX-2)
Cyclooxygenases (COX-1 and -2) are rate-limiting enzymes in the production of prostaglandins. COX-1 expression is generally constitutive, whereas COX-2 is usually induced by stimuli involved in inflammatory responses (35). Overexpression of COX-2 in transgenic mice using a bovine keratin-5 promoter caused chronic pancreatitis-like state by 3 months of age. By 6 to 8 months, strongly dysplastic features suggestive of pancreatic ductal adenocarcinoma emerged in the metaplastic ducts. The abnormal pancreatic phenotype can be completely prevented by maintaining mice on a diet containing celecoxib, a well-characterized COX-2 inhibitor (22). In contrast, COX-2 over expression in pancreatic acinar cells increased pancreas size but did not affect histology within 5-6 months. After 8 months, COX-2-expressing mice developed chronic inflammation and numerous pancreatic cysts (ductal ectasia). None of the COX-2 expressing mice developed PanINs or tumors within 1 year. However, Cox-2 and mutant KRas co-expression caused dramatic inflammation resembling severe chronic pancreatitis and abundant PanINs (24). In line with this study, COX-2 but not COX-1 deficiency attenuated the severity of AP (34).
Interleukin-1 Beta (IL-1β)
Pancreatitis is associated with the increase of numerous cytokine/chemokines (60). A transgenic approach can be used to study the function of these factors. For example, IL-1β is a proinflammatory cytokine involved in many inflammation pathways. In a transgenic mouse model in which a rat elastase promoter drives the expression of human IL-1β, the pancreas was atrophied, and there was increased acinar proliferation and apoptosis, which is typical for chronic pancreatitis. Older mice displayed acinar-ductal metaplasia but did not develop neoplasia (82).
Lymphotoxin (LT) α and β
Lymphotoxin (LT)α and LTβ are cytokines of the TNF superfamily and are involved in the regulation of immunity. LTα and β mRNA levels were increased in pancreatic tissues from patients with AIP. Acinar-specific overexpression of LTαβ (Ela1-LTαβ) in mice led to an autoimmune disorder with various features similar to AIP. Chronic inflammation developed only in the pancreas but was sufficient to cause systemic autoimmunity. Acinar-specific overexpression of LTαβ did not cause autoimmunity in lymphocyte-deficient mice (113). This transgenic mouse model has the critical features of human AIP, including progressive pancreatitis and the formation of B- and T-cell zones that are reminiscent of tertiary lymphoid tissues. Most surprisingly, the overexpression of LT in the pancreas was sufficient to trigger a systemic autoimmune response that involved distant organs (5).
7. Other Models
Autophagy
Autophagy degrades intracellular protein aggregates and damaged or unneeded organelles through alysosome-driven process and recycles components vital for cell survival. There are three major autophagic pathways: chaperone-mediated autophagy, microautophagy, and macroautophagy (40). The major form of autophagy is macroautophagy.
Impaired autophagic flux can lead to the formation of large vacuoles due to the fusion of autophagic vacuoles. Accumulation of large vacuoles in acinar cells is a prominent feature of pancreatitis, and most of these vacuoles are predominantly autolysosomes accompanied by increased LC3-II and p62, which are signs of increased and defective autophagy (81). Impaired autophagy can cause accumulation of damaged mitochondria, resulting in decreased ATP production, reactive oxygen species (ROS) overproduction , inflammasome activation, and ultimately cell death (42). Impaired autophagy also induces the accumulation of aggregates that containing p62, a signaling hub for oxidative stress and NF-κB pathways (91). In addition, autophagic dysfunction mediates the accumulation of active trypsin in acinar cells, another key mechanism of pancreatitis (81).
The assembly of Atg5-Atg12-Atg16 and the Atg5-Atg12-Atg16-mediated conjugation of LC3 I with phosphatidylethanolamine is key to the formation of autophagosomes. Therefore, the absence of Atg5 blocks autophagic processing, leading to the accumulation of protein aggregates and damaged organelles (85). The role of Atg5 was recently studied in pancreas-specific Atg5 -deficient mice by crossing Atg5flox/flox to Ptf1aCre mice (27). Pancreata were normal in 1-week-old Atg5 deficient mice. However, the loss of Atg5 resulted in edematous and enlarged pancreata by 4 weeks of age. With increasing age, the pancreata became atrophic, and the mice lost a significant amount of body weight. Histologically, acinar cells exhibited early cytoplasmic vacuolization and acquired a hypertrophic phenotype. Evidence of incremental fibrosis, pancreatic stellate cell activation, duct-like structures, inflammation, proliferation, apoptosis, and necrosis were evident. Blocked autophagic degradation was also evidenced by the accumulation of improperly formed autophagosomes containing various cellular constituents. Increased serum lipase activities, increased trypsin, and cathepsin B activities were found in pancreatic tissue. Defective pancreatic autophagy leads to the accumulation of p62, damaged mitochondria, increased ROS, and terminal ER stress. ROS and p62 activate Nrf2/Nqo1/p53 signaling thereby exacerbating cellular stress, necrosis/apoptosis, inflammation, and fibrosis. Surprisingly, the development of CP was much less pronounced in female mice than in male mice. Antioxidative treatment prevents the progression of CP in male mice (27). This study represents the first detailed analysis of the effects of genetic ablation of a key autophagy mediator in pancreas (44).
Keratin K8 and K18
Keratin K8 and K18 are the major components of the intermediate-filament cytoskeleton of simple epithelia. Transgenic mice expressing the human K8 (hk8) gene led to a moderate keratin-content increase in the epithelia (18). These mice displayed progressive exocrine pancreas alterations, including dysplasia, loss of acinar architecture, redifferentiation of acinar cells to ductal cells, inflammation, fibrosis, and substitution of exocrine tissue by adipose tissue, as well as increased cell proliferation and apoptosis. These results indicate that simple epithelial keratins play a relevant role in the regulation of exocrine pancreas homeostasis (19). In contrast, another study showed that K8- and K18-overexpressing pancreata were histologically similar to those in WT mice, whereas K8/K18 pancreata displayed age-enhanced vacuolization and atrophy of the exocrine pancreas. Zymogen granules in K8/K18 pancreata were 50% smaller and more dispersed than their normal apical concentration (125).
Liver X Receptors (LXR)
LXRα and LXRβ are nuclear receptors belonging to the ligand-activated transcription factor superfamily and play a key role in controlling lipid and glucose metabolism. The ligands for LXRs are 22-hydroxycholesterol, 24(S)-hydroxycholesterol, 24(S), 25-epoxycholesterol, and 27-hydroxycholesterol, which at physiological concentrations bind to and activate these receptors. LXRβ-/- mice showed pancreatic exocrine insufficiency with reduced serum levels of amylase and lipase, and the pancreas pathology indicated chronic inflammatory infiltration and an increased apoptosis without compensatory proliferation in the ductal epithelium (3, 36).
The Serum Response Factor (SRF)
SRF is a transcription factor regulating many immediate early genes and has been implicated in the control of differentiation, growth, and cell death. Using pancreatic specific disruption of this gene, it has been shown that SRF is indispensable for pancreatic ontogenesis; and after weaning, these mice developed profound pancreatitis. At the age of 4 months the exocrine pancreas had completely disappeared in most animals and was replaced by adipose tissue. Interestingly, the organization and function of the endocrine islets of Langerhans remained well preserved even though the PDX-Cre also targets the deletion of SRF in these cells (84).
Cilia
Defects in cilia formation or function have been implicated in several human genetic diseases, including polycystic kidney disease (PKD). Pancreatic lesions are found in approximately 10% of PKD patients, suggesting a connection between cilia defects and pancreatic pathologies. Kif3a, the gene encoding for a subunit of the kinesin-2 complex that is essential for cilia formation, was conditionally inactivated in pancreatic epithelia using PDX-Cre. The pancreata of these mutant mice displayed a loss of acinar cells shortly after birth and an acinar-to-ductal metaplasia and periductal fibrosis by 2 weeks after birth. At 12 weeks, the acinar cells were replaced by adipose tissue. At 6 months, the pancreata in these mice were composed of cysts that enlarged over time (17, 112).
8. Summary
The recent use of genetically engineered mice in pancreatic research has greatly improved our understanding of the molecular mechanisms of pancreatitis. Genetic approaches have also assisted the development of clinically relevant models of pancreatitis. In general, the genetic approaches are believed to be more specific than pharmacological compounds for targeting a particular pathway. However, caution should be used when interpreting the results from genetic studies: expression of a transgene at an irrelevant levels may give rise to artificial effects or no phenotype; expression of an irrelevant gene (eg. toxin) may not reflect the pathogenesis in human conditions; genetic deletion of a gene may also cause paradoxical effects because many genes (ER stress related genes, NF-κB) are important in the pathogenesis of inflammation but are also critical for homeostasis; Due to species-specific gene functions, different phenotypic patterns or no phenotype may develop when mouse counterparts are targeted; “off target” effects should also be considered because a single gene may regulate many pathways. In addition, pancreatitis is caused by multifaceted mechanisms (62), most of the genetically engineered pancreatitis models created by targeting one gene may not be appropriate for testing general preventive and therapeutic interventions. It should also be noted that pancreatitis initiation, progression and regression involve the interaction between pancreatic parenchymal cells with inflammatory cells (CD11c-Cre, LysM-Cre, F4/80-Cre, etc), stellate cells (SMA-Cre), and endothelial cells (VECadherin-CreERT) that can also be targeted. Using these cell-specific Cre to target gene expressions in the specific cell population are also of great interest in pancreatitis research.
Acknowledgements
The project described was supported by P50 CA102701 and K12 CA90628 from the National Cancer Institute and W81XWH-15-1-0257 from the Department of Defense. This publication was also supported by the CTSA Grant UL1 TR000135 from the National Center for Advancing Translational Sciences (NCATS), a component of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Department of Defense. We also thank Ms. Kelly E. Viola, ELS, for critical editing.
9. References
- Adolph TE,Niederreiter L,Blumberg RS and Kaser A. Endoplasmic reticulum stress and inflammation. Digestive Dis 30(4): 341-346,2012. PMID: 22796794.
- Aguirre AJ,Bardeesy N,Sinha M,Lopez L,Tuveson DA,Horner J, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev 17(24): 3112-3126,2003. PMID: 14681207.
- Alberti S,Schuster G,Parini P,Feltkamp D,Diczfalusy U,Rudling M, et al. Hepatic cholesterol metabolism and resistance to dietary cholesterol in LXRbeta-deficient mice. J Clin Invest 107(5): 565-573,2001. PMID: 11238557.
- Aleksic T,Baumann B,Wagner M,Adler G,Wirth T and Weber CK. Cellular immune reaction in the pancreas is induced by constitutively active IkappaB kinase-2. Gut 56(2): 227-236,2007. PMID: 16870717.
- Algul H and Chari ST. Lymphotoxin in the pathogenesis of autoimmune pancreatitis: a new player in the field. Gastroenterology 143(5): 1147-1150,2012. PMID: 23000229.
- Algul H,Treiber M,Lesina M,Nakhai H,Saur D,Geisler F, et al. Pancreas-specific RelA/p65 truncation increases susceptibility of acini to inflammation-associated cell death following cerulein pancreatitis. J Clin Invest 117(6): 1490-1501,2007. PMID: 17525802.
- Antonarakis SE. Recommendations for a nomenclature system for human gene mutations. Nomenclature Working Group. Human mutation 11(1): 1-3,1998. PMID: 9450896.
- Archer H,Jura N,Keller J,Jacobson M and Bar-Sagi D. A mouse model of hereditary pancreatitis generated by transgenic expression of R122H trypsinogen. Gastroenterology 131(6): 1844-1855,2006. PMID: 17087933.
- Athwal T,Huang W,Mukherjee R,Latawiec D,Chvanov M,Clarke R, et al. Expression of human cationic trypsinogen (PRSS1) in murine acinar cells promotes pancreatitis and apoptotic cell death. Cell Death & Dis 5: e1165,2014. PMID: 24722290.
- Awano H,Lee T,Yagi M,Masamune A,Kume K,Takeshima Y, et al. Childhood-onset hereditary pancreatitis with mutations in the CT gene and SPINK1 gene. Pediatrics international 55(5): 646-649,2013. PMID: 24134754.
- Baumann B,Wagner M,Aleksic T,von Wichert G,Weber CK,Adler G, et al. Constitutive IKK2 activation in acinar cells is sufficient to induce pancreatitis in vivo. J Clin Invest 117(6): 1502-1513,2007. PMID: 17525799.
- Beg AA,Sha WC,Bronson RT and Baltimore D. Constitutive NF-kappa B activation, enhanced granulopoiesis, and neonatal lethality in I kappa B alpha-deficient mice. Genes Dev 9(22): 2736-2746,1995. PMID: 7590249.
- Bialek R,Willemer S,Arnold R and Adler G. Evidence of intracellular activation of serine proteases in acute cerulein-induced pancreatitis in rats. Scand J Gastroenterol 26(2): 190-196,1991. PMID: 1707179.
- Blackstone M. Premature trypsin activation in hereditary pancreatitis. Gastroenterology 115(3): 796-799,1998. PMID: 9742004.
- Boomershine CS,Chamberlain A,Kendall P,Afshar-Sharif AR,Huang H,Washington MK, et al. Autoimmune pancreatitis results from loss of TGFbeta signalling in S100A4-positive dendritic cells. Gut 58(9): 1267-1274,2009. PMID: 19625278.
- Bottinger EP,Jakubczak JL,Roberts IS,Mumy M,Hemmati P,Bagnall K, et al. Expression of a dominant-negative mutant TGF-beta type II receptor in transgenic mice reveals essential roles for TGF-beta in regulation of growth and differentiation in the exocrine pancreas. EMBO J 16(10): 2621-2633,1997. PMID: 9184209.
- Cano DA,Sekine S and Hebrok M. Primary cilia deletion in pancreatic epithelial cells results in cyst formation and pancreatitis. Gastroenterology 131(6): 1856-1869,2006. PMID: 17123526.
- Casanova L,Bravo A,Were F,Ramirez A,Jorcano JJ and Vidal M. Tissue-specific and efficient expression of the human simple epithelial keratin 8 gene in transgenic mice. J Cell Sci 108 ( Pt 2): 811-820,1995. PMID: 7539440.
- Casanova ML,Bravo A,Ramirez A,Morreale de Escobar G,Were F,Merlino G, et al. Exocrine pancreatic disorders in transgenic mice expressing human keratin 8. J Clin Invest 103(11): 1587-1595,1999. PMID: 10359568.
- Chen JM and Ferec C. Genes, cloned cDNAs, and proteins of human trypsinogens and pancreatitis-associated cationic trypsinogen mutations. Pancreas 21(1): 57-62,2000. PMID: 10881933.
- Chiari H. Über die Selbstverdauung des menschlichen Pankreas. Zeitschrift für Heilkunde 1896;17 69-96.
- Colby JK,Klein RD,McArthur MJ,Conti CJ,Kiguchi K,Kawamoto T, et al. Progressive metaplastic and dysplastic changes in mouse pancreas induced by cyclooxygenase-2 overexpression. Neoplasia 10(8): 782-796,2008. PMID: 18670639.
- Comfort MW and Steinberg AG. Pedigree of a family with hereditary chronic relapsing pancreatitis. Gastroenterology 21(1): 54-63,1952. PMID: 14926813.
- Daniluk J,Liu Y,Deng D,Chu J,Huang H,Gaiser S, et al. An NF-kappaB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J Clin Invest 122(4): 1519-1528,2012. PMID: 22406536.
- Dawra R,Sah RP,Dudeja V,Rishi L,Talukdar R,Garg P, et al. Intra-acinar trypsinogen activation mediates early stages of pancreatic injury but not inflammation in mice with acute pancreatitis. Gastroenterology 141(6): 2210-2217 e2212,2011. PMID: 21875495.
- Delhase M,Hayakawa M,Chen Y and Karin M. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science 284(5412): 309-313,1999. PMID: 10195894.
- Diakopoulos KN,Lesina M,Wormann S,Song L,Aichler M,Schild L, et al. Impaired autophagy induces chronic atrophic pancreatitis in mice via sex- and nutrition-dependent processes. Gastroenterology 148(3): 626-638 e617,2015. PMID: 25497209.
- DiDonato JA,Mercurio F and Karin M. NF-kappaB and the link between inflammation and cancer. Immunological Rev 246(1): 379-400,2012. PMID: 22435567.
- Dimagno MJ,Lee SH,Hao Y,Zhou SY,McKenna BJ and Owyang C. A proinflammatory, antiapoptotic phenotype underlies the susceptibility to acute pancreatitis in cystic fibrosis transmembrane regulator (-/-) mice. Gastroenterology 129(2): 665-681,2005. PMID: 16083720.
- DiMagno MJ,Lee SH,Owyang C and Zhou SY. Inhibition of acinar apoptosis occurs during acute pancreatitis in the human homologue DeltaF508 cystic fibrosis mouse. Am J Physiol Gastrointest Liver Physiol 299(2): G400-412,2010. PMID: 20522641.
- Dorin JR,Farley R,Webb S,Smith SN,Farini E,Delaney SJ, et al. A demonstration using mouse models that successful gene therapy for cystic fibrosis requires only partial gene correction. Gene Therapy 3(9): 797-801,1996. PMID: 8875228.
- Dufey E,Sepulveda D,Rojas-Rivera D and Hetz C. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 1. An overview. Am J P. Cell physiology 307(7): C582-594,2014. PMID: 25143348.
- Durie PR,Kent G,Phillips MJ and Ackerley CA. Characteristic multiorgan pathology of cystic fibrosis in a long-living cystic fibrosis transmembrane regulator knockout murine model. Am J Pathol 164(4): 1481-1493,2004. PMID: 15039235.
- Ethridge RT,Chung DH,Slogoff M,Ehlers RA,Hellmich MR,Rajaraman S, et al. Cyclooxygenase-2 gene disruption attenuates the severity of acute pancreatitis and pancreatitis-associated lung injury. Gastroenterology 123(4): 1311-1322,2002. PMID: 12360491.
- FitzGerald GA. COX-2 and beyond: Approaches to prostaglandin inhibition in human disease. Nature reviews. Drug Discovery 2(11): 879-890,2003. PMID: 14668809.
- Gabbi C,Kim HJ,Hultenby K,Bouton D,Toresson G,Warner M, et al. Pancreatic exocrine insufficiency in LXRbeta-/- mice is associated with a reduction in aquaporin-1 expression. Proc Natl Acad Sci U S A 105(39): 15052-15057,2008. PMID: 18806227.
- Gaiser S,Daniluk J,Liu Y,Tsou L,Chu J,Lee W, et al. Intracellular activation of trypsinogen in transgenic mice induces acute but not chronic pancreatitis. Gut 60(10): 1379-1388,2011. PMID: 21471572.
- Garg AD,Kaczmarek A,Krysko O,Vandenabeele P,Krysko DV and Agostinis P. ER stress-induced inflammation: does it aid or impede disease progression? Trends Mol Med 18(10): 589-598,2012. PMID: 22883813.
- Gilliland L and Steer ML. Effects of ethionine on digestive enzyme synthesis and discharge by mouse pancreas. Am J Physiol Gastrointest Liver Physiol 239(5): G418-426,1980. PMID: 6159794.
- Glick D,Barth S and Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol 221(1): 3-12,2010. PMID: 20225336.
- Grady T,Liang P,Ernst SA and Logsdon CD. Chemokine gene expression in rat pancreatic acinar cells is an early event associated with acute pancreatitis. Gastroenterology 113(6): 1966-1975,1997. PMID: 9394737.
- Green DR,Galluzzi L and Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 333(6046): 1109-1112,2011. PMID: 21868666.
- Gukovsky I and Gukovskaya A. Nuclear factor-kappaB in pancreatitis: Jack-of-all-trades, but which one is more important? Gastroenterology 144(1): 26-29,2013. PMID: 23164573.
- Gukovsky I and Gukovskaya AS. Impaired autophagy triggers chronic pancreatitis: lessons from pancreas-specific atg5 knockout mice. Gastroenterology 148(3): 501-505,2015. PMID: 25613315.
- Gukovsky I,Pandol SJ and Gukovskaya AS. Organellar dysfunction in the pathogenesis of pancreatitis. Antioxidants & Redox Signaling 15(10): 2699-2710,2011. PMID: 21834686.
- Hacker H and Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE 2006(357): re13,2006. PMID: 17047224.
- Hahm KB,Im YH,Lee C,Parks WT,Bang YJ,Green JE, et al. Loss of TGF-beta signaling contributes to autoimmune pancreatitis. J Clin Invest 105(8): 1057-1065,2000. PMID: 10772650.
- Han B,Ji B and Logsdon CD. CCK independently activates intracellular trypsinogen and NF-kappaB in rat pancreatic acinar cells. Am J Physiol Cell Physiol 280(3): C465-472,2001. PMID: 11171565.
- Harding HP,Zeng H,Zhang Y,Jungries R,Chung P,Plesken H, et al. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol Cell 7(6): 1153-1163,2001. PMID: 11430819.
- Herrmann O,Baumann B,de Lorenzi R,Muhammad S,Zhang W,Kleesiek J, et al. IKK mediates ischemia-induced neuronal death. Nature Medicine 11(12): 1322-1329,2005. PMID: 16286924.
- Hess DA,Humphrey SE,Ishibashi J,Damsz B,Lee AH,Glimcher LH, et al. Extensive pancreas regeneration following acinar-specific disruption of Xbp1 in mice. Gastroenterology 141(4): 1463-1472,2011. PMID: 21704586.
- Hingorani SR,Petricoin EF,Maitra A,Rajapakse V,King C,Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4(6): 437-450,2003. PMID: 14706336.
- Hinz M and Scheidereit C. The IkappaB kinase complex in NF-kappaB regulation and beyond. EMBO Reports 15(1): 46-61,2014. PMID: 24375677.
- Hoffmann A and Baltimore D. Circuitry of nuclear factor kappaB signaling. Immunol Rev 210: 171-186,2006. PMID: 16623771.
- Hu Y,Baud V,Delhase M,Zhang P,Deerinck T,Ellisman M, et al. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science 284(5412): 316-320,1999. PMID: 10195896.
- Huang H,Daniluk J,Liu Y,Chu J,Li Z,Ji B, et al. Oncogenic K-Ras requires activation for enhanced activity. Oncogene 33(4): 532-535,2014. PMID: 23334325.
- Huang H,Liu Y,Daniluk J,Gaiser S,Chu J,Wang H, et al. Activation of nuclear factor-kappaB in acinar cells increases the severity of pancreatitis in mice. Gastroenterology 144(1): 202-210,2013. PMID: 23041324.
- Iida K,Li Y,McGrath BC,Frank A and Cavener DR. PERK eIF2 alpha kinase is required to regulate the viability of the exocrine pancreas in mice. BMC Cell Biol 8: 38,2007. PMID: 17727724.
- Imrie JR,Fagan DG and Sturgess JM. Quantitative evaluation of the development of the exocrine pancreas in cystic fibrosis and control infants. Am J Pathol 95(3): 697-708,1979. PMID: 453330.
- Ji B,Chen XQ,Misek DE,Kuick R,Hanash S,Ernst S, et al. Pancreatic gene expression during the initiation of acute pancreatitis: identification of EGR-1 as a key regulator. Physiol Genomics 14(1): 59-72,2003. PMID: 12709512.
- Ji B,Gaiser S,Chen X,Ernst SA and Logsdon CD. Intracellular trypsin induces pancreatic acinar cell death but not NF-kappaB activation. J Biol Chem 284(26): 17488-17498,2009. PMID: 19383608.
- Ji B and Logsdon CD. Digesting new information about the role of trypsin in pancreatitis. Gastroenterology 141(6): 1972-1975,2011. PMID: 22033179.
- Ji B,Song J,Tsou L,Bi Y,Gaiser S,Mortensen R, et al. Robust acinar cell transgene expression of CreErT via BAC recombineering. Genesis 46(8): 390-395,2008. PMID: 18693271.
- Ji B,Tsou L,Wang H,Gaiser S,Chang DZ,Daniluk J, et al. Ras activity levels control the development of pancreatic diseases. Gastroenterology 137(3): 1072-1082, 1082 e1071-1076,2009. PMID: 19501586.
- Kazal LA,Spicer DS and Brahinsky RA. Isolation of a crystalline trypsin inhibitor-anticoagulant protein from pancreas. J Am Chem Soc 70(9): 3034-3040,1948. PMID: 18882536.
- Kistner A,Gossen M,Zimmermann F,Jerecic J,Ullmer C,Lubbert H, et al. Doxycycline-mediated quantitative and tissue-specific control of gene expression in transgenic mice. Proc Natl Acad Sci U S A 93(20): 10933-10938,1996. PMID: 8855286.
- Klement JF,Rice NR,Car BD,Abbondanzo SJ,Powers GD,Bhatt PH, et al. IkappaBalpha deficiency results in a sustained NF-kappaB response and severe widespread dermatitis in mice. Mol Cell Biol 16(5): 2341-2349,1996. PMID: 8628301.
- Koike H,Steer ML and Meldolesi J. Pancreatic effects of ethionine: blockade of exocytosis and appearance of crinophagy and autophagy precede cellular necrosis. Am J Physiol Gastrointest Liver Physiol 242(4): G297-307,1982. PMID: 7065251.
- Kubisch CH and Logsdon CD. Endoplasmic reticulum stress and the pancreatic acinar cell. Expert Review Gastroenterol Hepatol 2(2): 249-260,2008. PMID: 19072360.
- LaRusch J,Solomon S and Whitcomb DC. Pancreatitis Overview. GeneReviews(R). RA Pagon, MP Adam, HH Ardinger, SE Wallace, A Amemiya, LJH Bean et al. Seattle (WA), 1993.
- Le Bodic L,Bignon JD,Raguenes O,Mercier B,Georgelin T,Schnee M, et al. The hereditary pancreatitis gene maps to long arm of chromosome 7. Hum Mol Genet 5(4): 549-554,1996. PMID: 8845851.
- Leach SD,Modlin IM,Scheele GA and Gorelick FS. Intracellular activation of digestive zymogens in rat pancreatic acini. Stimulation by high doses of cholecystokinin. J Clin Invest 87(1): 362-366,1991. PMID: 1985109.
- Lee MS,Gu D,Feng L,Curriden S,Arnush M,Krahl T, et al. Accumulation of extracellular matrix and developmental dysregulation in the pancreas by transgenic production of transforming growth factor-beta 1. Am J Pathol 147(1): 42-52,1995. PMID: 7604884.
- Li N,Wu X,Holzer RG,Lee JH,Todoric J,Park EJ, et al. Loss of acinar cell IKKalpha triggers spontaneous pancreatitis in mice. J Clin Invest 123(5): 2231-2243,2013. PMID: 23563314.
- Liu B,Xia X,Zhu F,Park E,Carbajal S,Kiguchi K, et al. IKKalpha is required to maintain skin homeostasis and prevent skin cancer. Cancer Cell 14(3): 212-225,2008. PMID: 18772111.
- Logsdon CD and Ji B. Ras activity in acinar cells links chronic pancreatitis and pancreatic cancer. Clin Gastroenterol Hepatol 7(11 Suppl): S40-43,2009. PMID: 19896097.
- Lugea A,Tischler D,Nguyen J,Gong J,Gukovsky I,French SW, et al. Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology 140(3): 987-997,2011. PMID: 21111739.
- Lugea A,Waldron RT and Pandol SJ. Pancreatic adaptive responses in alcohol abuse: Role of the unfolded protein response. Pancreatology,2015. PMID: 25736240.
- Luo S,Mao C,Lee B and Lee AS. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol Cell Biol 26(15): 5688-5697,2006. PMID: 16847323.
- Marchbank T,Freeman TC and Playford RJ. Human pancreatic secretory trypsin inhibitor. Distribution, actions and possible role in mucosal integrity and repair. Digestion 59(3): 167-174,1998. PMID: 9643675.
- Mareninova OA,Hermann K,French SW,O'Konski MS,Pandol SJ,Webster P, et al. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J Clin Invest 119(11): 3340-3355,2009. PMID: 19805911.
- Marrache F,Tu SP,Bhagat G,Pendyala S,Osterreicher CH,Gordon S, et al. Overexpression of interleukin-1beta in the murine pancreas results in chronic pancreatitis. Gastroenterology 135(4): 1277-1287,2008. PMID: 18789941.
- Mercurio F,Zhu H,Murray BW,Shevchenko A,Bennett BL,Li J, et al. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 278(5339): 860-866,1997. PMID: 9346484.
- Miralles F,Hebrard S,Lamotte L,Durel B,Gilgenkrantz H,Li Z, et al. Conditional inactivation of the murine serum response factor in the pancreas leads to severe pancreatitis. Lab Invest 86(10): 1020-1036,2006. PMID: 16894357.
- Mizushima N and Komatsu M. Autophagy: renovation of cells and tissues. Cell 147(4): 728-741,2011. PMID: 22078875.
- Napetschnig J and Wu H. Molecular basis of NF-kappaB signaling. Ann Rev Biophys 42: 443-468,2013. PMID: 23495970.
- Nathan JD,Romac J,Peng RY,Peyton M,Macdonald RJ and Liddle RA. Transgenic expression of pancreatic secretory trypsin inhibitor-I ameliorates secretagogue-induced pancreatitis in mice. Gastroenterology 128(3): 717-727,2005. PMID: 15765407.
- Nemeth BC and Sahin-Toth M. Human cationic trypsinogen (PRSS1) variants and chronic pancreatitis. Am J Physiol Gastrointest Liver Physiol 306(6): G466-473,2014. PMID: 24458023.
- Neuhofer P,Liang S,Einwachter H,Schwerdtfeger C,Wartmann T,Treiber M, et al. Deletion of IkappaBalpha activates RelA to reduce acute pancreatitis in mice through up-regulation of Spi2A. Gastroenterology 144(1): 192-201,2013. PMID: 23041330.
- Neuschwander-Tetri BA,Fimmel CJ,Kladney RD,Wells LD and Talkad V. Differential expression of the trypsin inhibitor SPINK3 mRNA and the mouse ortholog of secretory granule protein ZG-16p mRNA in the mouse pancreas after repetitive injury. Pancreas 28(4): e104-111,2004. PMID: 15097871.
- Nezis IP and Stenmark H. p62 at the interface of autophagy, oxidative stress signaling, and cancer. Antioxidants & Redox Signaling 17(5): 786-793,2012. PMID: 22074114.
- Oeckinghaus A and Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harbor Perspectives Biology 1(4): a000034,2009. PMID: 20066092.
- Ohmuraya M,Hirota M,Araki K,Baba H and Yamamura K. Enhanced trypsin activity in pancreatic acinar cells deficient for serine protease inhibitor kazal type 3. Pancreas 33(1): 104-106,2006. PMID: 16804421.
- Ohmuraya M,Hirota M,Araki M,Mizushima N,Matsui M,Mizumoto T, et al. Autophagic cell death of pancreatic acinar cells in serine protease inhibitor Kazal type 3-deficient mice. Gastroenterology 129(2): 696-705,2005. PMID: 16083722.
- Ohmuraya M and Yamamura K. Roles of serine protease inhibitor Kazal type 1 (SPINK1) in pancreatic diseases. Experimental animals / Japanese Association for Laboratory Animal Science 60(5): 433-444,2011. PMID: 22041280.
- Ostedgaard LS,Meyerholz DK,Chen JH,Pezzulo AA,Karp PH,Rokhlina T, et al. The DeltaF508 mutation causes CFTR misprocessing and cystic fibrosis-like disease in pigs. Science Translational Medicine 3(74): 74ra24,2011. PMID: 21411740.
- Paju A and Stenman UH. Biochemistry and clinical role of trypsinogens and pancreatic secretory trypsin inhibitor. Critical reviews in clinical laboratory sciences 43(2): 103-142,2006. PMID: 16517420.
- Pandya A,Blanton SH,Landa B,Javaheri R,Melvin E,Nance WE, et al. Linkage studies in a large kindred with hereditary pancreatitis confirms mapping of the gene to a 16-cM region on 7q. Genomics 38(2): 227-230,1996. PMID: 8954806.
- Peng B,Ling J,Lee AJ,Wang Z,Chang Z,Jin W, et al. Defective feedback regulation of NF-kappaB underlies Sjogren's syndrome in mice with mutated kappaB enhancers of the IkappaBalpha promoter. Proc Natl Acad Sci U S A 107(34): 15193-15198,2010. PMID: 20696914.
- Petersen OH,Gerasimenko OV,Tepikin AV and Gerasimenko JV. Aberrant Ca(2+) signalling through acidic calcium stores in pancreatic acinar cells. Cell Calcium 50(2): 193-199,2011. PMID: 21435718.
- Rane SG,Lee JH and Lin HM. Transforming growth factor-beta pathway: role in pancreas development and pancreatic disease. Cytokine & Growth factor 17(1-2): 107-119,2006. PMID: 16257256.
- Riordan JR,Rommens JM,Kerem B,Alon N,Rozmahel R,Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245(4922): 1066-1073,1989. PMID: 2475911.
- Rogers CS,Hao Y,Rokhlina T,Samuel M,Stoltz DA,Li Y, et al. Production of CFTR-null and CFTR-DeltaF508 heterozygous pigs by adeno-associated virus-mediated gene targeting and somatic cell nuclear transfer. J Clin Invest 118(4): 1571-1577,2008. PMID: 18324337.
- Rogers CS,Stoltz DA,Meyerholz DK,Ostedgaard LS,Rokhlina T,Taft PJ, et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 321(5897): 1837-1841,2008. PMID: 18818360.
- Romac JM,Ohmuraya M,Bittner C,Majeed MF,Vigna SR,Que J, et al. Transgenic expression of pancreatic secretory trypsin inhibitor-1 rescues SPINK3-deficient mice and restores a normal pancreatic phenotype. Am J Physiol Gastrointest Liver Physiol 298(4): G518-524,2010. PMID: 20110462.
- Romac JM,Shahid RA,Choi SS,Karaca GF,Westphalen CB,Wang TC, et al. Pancreatic Secretory Trypsin Inhibitor 1 Reduces the Severity of Chronic Pancreatitis in Mice Over-expressing Interleukin-1beta in the Pancreas. Am J Physiol Gastrointest Liver Physiol,2011. PMID: 22173919.
- Rothwarf DM and Karin M. The NF-kappa B activation pathway: a paradigm in information transfer from membrane to nucleus. Science's STKE : signal transduction knowledge environment 1999(5): RE1,1999. PMID: 11865184.
- Sah RP,Dawra RK and Saluja AK. New insights into the pathogenesis of pancreatitis. Curr Opinion Gastroenterol 29(5): 523-530,2013. PMID: 23892538.
- Sah RP,Garg P and Saluja AK. Pathogenic mechanisms of acute pancreatitis. Curr Opinion Gastroenterol 28(5): 507-515,2012. PMID: 22885948.
- Sahin-Toth M and Toth M. Gain-of-function mutations associated with hereditary pancreatitis enhance autoactivation of human cationic trypsinogen. Biochem Biophys Res Commun 278(2): 286-289,2000. PMID: 11097832.
- Sakata K,Ohmuraya M,Araki K,Suzuki C,Ida S,Hashimoto D, et al. Generation and analysis of serine protease inhibitor kazal type 3-cre driver mice. Experimental animals / Japanese Association for Laboratory Animal Science 63(1): 45-53,2014. PMID: 24521862.
- Schmid RM and Whitcomb DC. Genetically defined models of chronic pancreatitis. Gastroenterology 131(6): 2012-2015,2006. PMID: 17123529.
- Seleznik GM,Reding T,Romrig F,Saito Y,Mildner A,Segerer S, et al. Lymphotoxin beta receptor signaling promotes development of autoimmune pancreatitis. Gastroenterology 143(5): 1361-1374,2012. PMID: 22863765.
- Selig L,Sack U,Gaiser S,Kloppel G,Savkovic V,Mossner J, et al. Characterisation of a transgenic mouse expressing R122H human cationic trypsinogen. BMC Gastroenterol 6: 30,2006. PMID: 17069643.
- Sen R and Baltimore D. Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell 47(6): 921-928,1986. PMID: 3096580.
- Shen RR and Hahn WC. Emerging roles for the non-canonical IKKs in cancer. Oncogene 30(6): 631-641,2011. PMID: 21042276.
- Siegel RL,Miller KD and Jemal A. Cancer statistics, 2016. CA: a cancer journal for clinicians 66(1): 7-30,2016. PMID: 26742998.
- Solomon S and Whitcomb DC. Genetics of pancreatitis: an update for clinicians and genetic counselors. Curr Gastroenterol Reports 14(2): 112-117,2012. PMID: 22314809.
- Stephen AG,Esposito D,Bagni RK and McCormick F. Dragging ras back in the ring. Cancer Cell 25(3): 272-281,2014. PMID: 24651010.
- Sun SC,Ganchi PA,Ballard DW and Greene WC. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science 259(5103): 1912-1915,1993. PMID: 8096091.
- Sun X,Olivier AK,Yi Y,Pope CE,Hayden HS,Liang B, et al. Gastrointestinal pathology in juvenile and adult CFTR-knockout ferrets. Am J Pathol 184(5): 1309-1322,2014. PMID: 24637292.
- Sun X,Sui H,Fisher JT,Yan Z,Liu X,Cho HJ, et al. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J Clin Invest 120(9): 3149-3160,2010. PMID: 20739752.
- Sun X,Yan Z,Yi Y,Li Z,Lei D,Rogers CS, et al. Adeno-associated virus-targeted disruption of the CFTR gene in cloned ferrets. J Clin Invest 118(4): 1578-1583,2008. PMID: 18324338.
- Suyama K,Ohmuraya M,Hirota M,Ozaki N,Ida S,Endo M, et al. C/EBP homologous protein is crucial for the acceleration of experimental pancreatitis. Biochem Biophys Res Commun 367(1): 176-182,2008. PMID: 18166146.
- Toivola DM,Nakamichi I,Strnad P,Michie SA,Ghori N,Harada M, et al. Keratin overexpression levels correlate with the extent of spontaneous pancreatic injury. Am J Pathol 172(4): 882-892,2008. PMID: 18349119.
- Treiber M,Neuhofer P,Anetsberger E,Einwachter H,Lesina M,Rickmann M, et al. Myeloid, but not pancreatic, RelA/p65 is required for fibrosis in a mouse model of chronic pancreatitis. Gastroenterology 141(4): 1473-1485, 1485 e1471-1477,2011. PMID: 21763242.
- van Acker GJ,Perides G and Steer ML. Co-localization hypothesis: a mechanism for the intrapancreatic activation of digestive enzymes during the early phases of acute pancreatitis. World J Gastroenterol 12(13): 1985-1990,2006. PMID: 16610045.
- Verhelst K,Verstrepen L,Carpentier I and Beyaert R. IkappaB kinase epsilon (IKKepsilon): a therapeutic target in inflammation and cancer. Biochem Pharmacol 85(7): 873-880,2013. PMID: 23333767.
- Weber P,Keim V and Zimmer KP. Hereditary pancreatitis and mutation of the trypsinogen gene. Arch Dis Child 80(5): 473-474,1999. PMID: 10208958.
- Weiss FU. Pancreatic cancer risk in hereditary pancreatitis. Front Physiol 5: 70,2014. PMID: 24600409.
- Weng TI,Wu HY,Chen BL,Jhuang JY,Huang KH,Chiang CK, et al. C/EBP homologous protein deficiency aggravates acute pancreatitis and associated lung injury. World J Gastroenterol 19(41): 7097-7105,2013. PMID: 24222953.
- Whitcomb DC. Hereditary pancreatitis: new insights into acute and chronic pancreatitis. Gut 45(3): 317-322,1999. PMID: 10446089.
- Whitcomb DC,Gorry MC,Preston RA,Furey W,Sossenheimer MJ,Ulrich CD, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet 14(2): 141-145,1996. PMID: 8841182.
- Whitcomb DC,Preston RA,Aston CE,Sossenheimer MJ,Barua PS,Zhang Y, et al. A gene for hereditary pancreatitis maps to chromosome 7q35. Gastroenterology 110(6): 1975-1980,1996. PMID: 8964426.
- Wildi S,Kleeff J,Mayerle J,Zimmermann A,Bottinger EP,Wakefield L, et al. Suppression of transforming growth factor beta signalling aborts caerulein induced pancreatitis and eliminates restricted stimulation at high caerulein concentrations. Gut 56(5): 685-692,2007. PMID: 17135311.
- Wilke M,Buijs-Offerman RM,Aarbiou J,Colledge WH,Sheppard DN,Touqui L, et al. Mouse models of cystic fibrosis: phenotypic analysis and research applications. J Cyst Fibrosis 10 Suppl 2: S152-171,2011. PMID: 21658634.
- Willemer S,Bialek R and Adler G. Localization of lysosomal and digestive enzymes in cytoplasmic vacuoles in caerulein-pancreatitis. Histochemistry 94(2): 161-170,1990. PMID: 2358374.
- Wilschanski M and Novak I. The cystic fibrosis of exocrine pancreas. Cold Spring Harb Perspect Med 3(5): a009746,2013. PMID: 23637307.
- Witt H,Luck W,Hennies HC,Classen M,Kage A,Lass U, et al. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet 25(2): 213-216,2000. PMID: 10835640.
- Wyllie R. Hereditary pancreatitis. Am J Gastroenterol 92(7): 1079-1080,1997. PMID: 9219774.
- Ye R,Mareninova OA,Barron E,Wang M,Hinton DR,Pandol SJ, et al. Grp78 heterozygosity regulates chaperone balance in exocrine pancreas with differential response to cerulein-induced acute pancreatitis. Am J Pathol 177(6): 2827-2836,2010. PMID: 20971738.
- Yoo BM,Yeo M,Oh TY,Choi JH,Kim WW,Kim JH, et al. Amelioration of pancreatic fibrosis in mice with defective TGF-beta signaling. Pancreas 30(3): e71-79,2005. PMID: 15782092.
- Zhou L,Dey CR,Wert SE,DuVall MD,Frizzell RA and Whitsett JA. Correction of lethal intestinal defect in a mouse model of cystic fibrosis by human CFTR. Science 266(5191): 1705-1708,1994. PMID: 7527588.