
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2015.25
| Attachment | Size |
|---|---|
| 394.99 KB |
Abstract:
Acute pancreatitis, chronic pancreatitis, and pancreatic cancer are three major pancreatic diseases without any effective treatment. In this character, the latest knowledge regarding the association between these diseases and tobacco smoking is reviewed. Clinical evidence of the association is presented, followed by an overview of scientific evidence, and the potential pathways that mediate smoking-induced pancreatitis and pancreatic cancer.
1. Introduction
Multiple clinical studies have shown that smoking tobacco, particularly cigarettes, elevates the risk for developing pancreatic diseases such as pancreatitis and cancer (summarized in (3, 39)). Furthermore, risk increases as a function of the amount of tobacco consumed. Smoking tobacco has often been linked as a co-factor with alcohol abuse in predisposing to pancreatic disorders. However, the inclusion of smokers that do not drink alcohol in some of these studies has highlighted that cigarette smoking can also be considered an independent risk factor. Despite significant clinical advancements in this field, scientific data exploring how tobacco toxins affect the pancreas at the cellular level are scarce (39). In this review we summarize clinical and scientific knowledge regarding the effects tobacco has upon the pancreas, and how it may contribute to disease development and progression.
2. Role of Tobacco in Development of Pancreatic Disease: Pancreatitis
Clinical Evidence
An exact role for tobacco intake as a risk factor in pancreatitis has been difficult to determine as chronic tobacco consumption is frequently associated with chronic alcohol abuse. More than 80% of patients with alcoholic chronic pancreatitis (ACP) are smokers and tobacco has largely been considered to potentiate alcohol toxicity (17, 71, 87). A retrospective cohort study of ACP showed that cigarette smoking altered the average age at ACP diagnosis; in smokers diagnosis occurred 5 years earlier compared to nonsmokers, and was coupled with increased risk of pancreatic calcification (71). These findings were validated by a recent study, which also found a concentration-dependence between ACP course and levels of tobacco consumption (87). Tobacco intake was measured in ‘pack years’, defined as the number of cigarettes per day multiplied by the number of years of smoking divided by 20 (20 cigarettes/pack). At a 10 pack year threshold no differences in ACP outcome were observed.
At a 20 pack year threshold, diagnosis of ACP was observed earlier and patients had more frequent calcifications; at a 30 pack year threshold similar results were seen, along with increased pancreatic exocrine insufficiency.
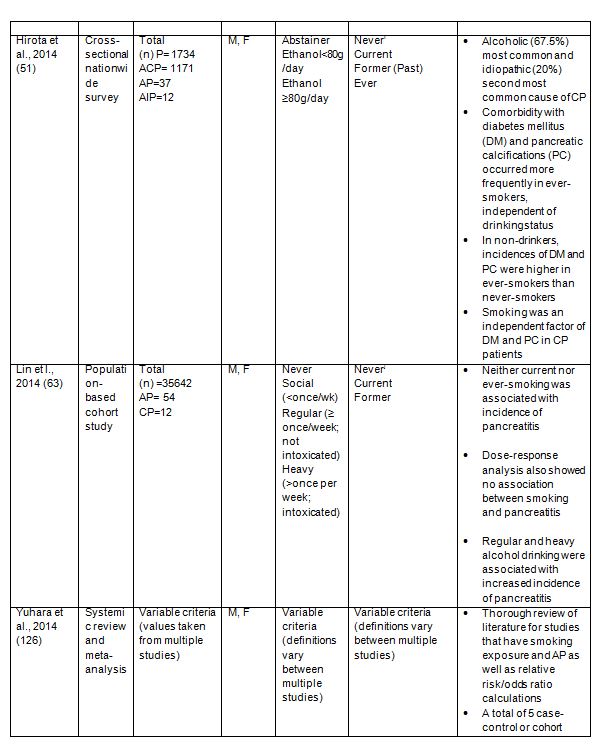
Although these studies imply tobacco use potentiates alcohol effects in pancreatitis convincing data has emerged from several case-control and cohort studies strongly supporting an independent association between smoking and pancreatitis (33, 51, 61, 63, 71, 92, 107, 108, 125, 126). The major findings from these studies are detailed in Table 1. All of these investigations conclude that smoking tobacco increases the risk for developing chronic pancreatitis (CP) independently of alcohol. For example, one U.S. study showed that, when compared with never-smokers, the relative risk (OR) for developing chronic pancreatitis in smokers with <12 pack years was 1.34 (95% CI 0.90-2.01), with 12-35 pack years it increased to 2.15 (95% CI 1.46-3.17) and with >35 pack years, the OR increased to 4.59 (95% CI 2.91-7.25). Further, in a stratified analysis, there was a direct correlation between the level of smoking and chronic pancreatitis for both men and women, Caucasians and “ever drinkers” (lifetime consumption of >20 alcoholic drinks) but not in the Black/African American population. While there was a trend toward increased risk in the African American community, the OR confidence intervals also increased, perhaps owing to the small number of subjects (123). Data arising from the Iowa Women’s Health Study, further supports a link between smoking levels and OR for CP; heavy smoking (40+ vs. 0 pack years) was associated with a two-fold increase in OR for CP, regardless of alcohol use (83). The fact that the study was directed at women equal to or greater than 65 years in age also suggests smoking may be a specific factor for developing CP in older women.
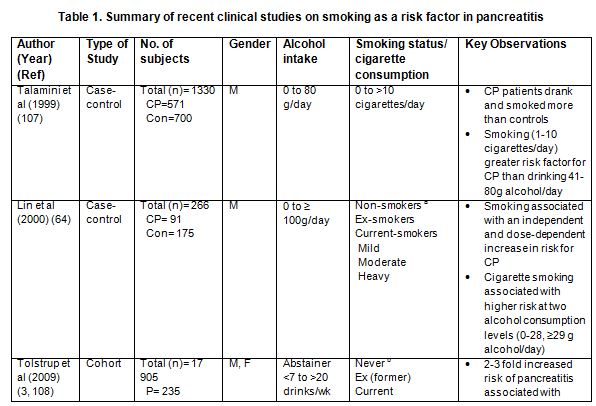


In Japan, a nationwide survey was conducted to clarify the epidemiological features of patients with CP (51). As it was a cross-sectional nationwide survey without healthy controls, it was not estimated whether smoking alone constituted a risk for the onset of CP. However the survey clearly showed that smoking tobacco increased the occurrence of clinical features associated with the disease. In this study, the incidence of comorbidity with diabetes mellitus and pancreatic calcifications increased significantly in the “never drinking but ever smoking” CP patients when compared to the “neither drinking nor smoking” CP patients. These findings imply that smoking poses a risk for developing CP complications independently of alcohol consumption.
Collectively, evidence from these clinical studies supports a dose-dependent association between smoking and chronic pancreatitis; however, a similar association with acute pancreatitis (AP) has also been revealed (18, 108, 126). In a Danish study with a mean follow-up of 20.2 years, a link between smoking and increased risk of AP was observed, independent of alcohol consumption. Another novel finding from this study was the risk for developing acute pancreatitis in former-smokers was elevated (1.7; 95% CI 1.0-2.7), compared to “never smokers” (108). However, this study did not account for the level of smoking by former-smokers (e.g. mild, moderate or heavy) or the extent of smoking abstinence, both of which could alter risk. A subsequent investigation centered on these factors and has exposed duration of smoking, rather than smoking intensity, as the reason for higher risk in this patient group. It was found that two decades after smoking cessation the relative risk (RR) was reduced to levels consistent with that seen in never-smokers (RR 1.20, 95% CI 0.66-2.15) (92). Although smoking cessation varied the risk for acute pancreatitis, another study found that no significant risk was associated with former smoking in terms of chronic pancreatitis (OR 0.40, 95% CI 0.14-1.18) (61). In addition to the Danish and American studies, a population-based cross-sectional study of the elderly Chinese population has linked tobacco consumption with the risk of AP, particularly in those who have smoked at least 15 pack years (125). Furthermore, systematic review and meta-analyses, which include many of the studies detailed here, find conclusively that current smoking and former smoking are firmly connected with elevated risk for AP (6, 126). One single-center prospective, cohort study of the natural history of AP, found that smoking was a dose-dependent risk factor for recurrent acute pancreatitis (RAP) (18). Furthermore they validated the association of cigarette smoking with CP. Therefore it seems likely that increased levels and/or duration of tobacco consumption could lead to repeat bouts of AP which ultimately evolve into CP.
It should be noted that one population-based cohort study in Taiwan did not find any evidence linking cigarette smoking and the incidence of pancreatitis, although they documented a dose-dependent association between alcohol abuse and pancreatitis (63). These findings are in sharp contrast to the overwhelming data supporting an independent effect of smoking and pancreatitis development (33, 51, 61, 63, 71, 92, 107, 108, 125, 126). In this study the authors reason that potential racial differences in nicotine metabolism and susceptibility to smoking between the Taiwanese populations they examined versus ethnically different populations from the other investigations, may account for the discrepancy. However, their study may not have been sufficiently powered to detect a modest association between smoking and pancreatitis, given that a small patient number developed pancreatitis, follow-up was relatively short, and alcohol and tobacco consumption in Taiwan was much lower compared with Western populations.
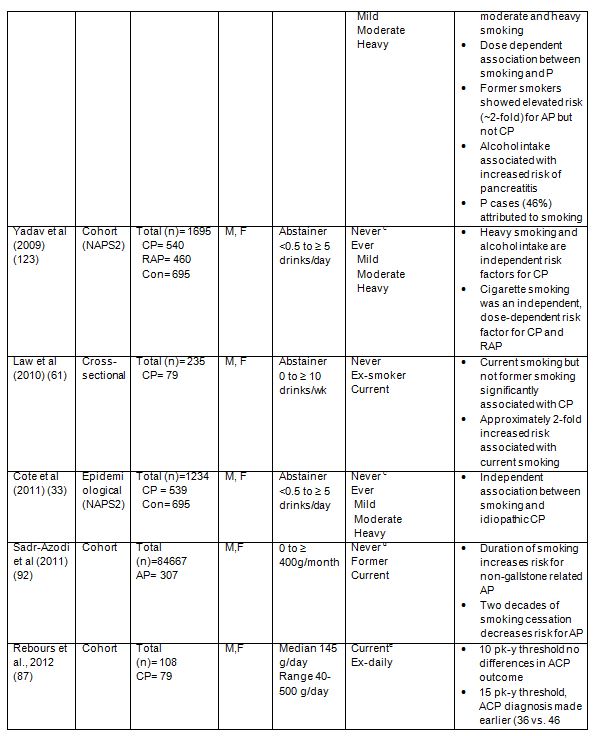
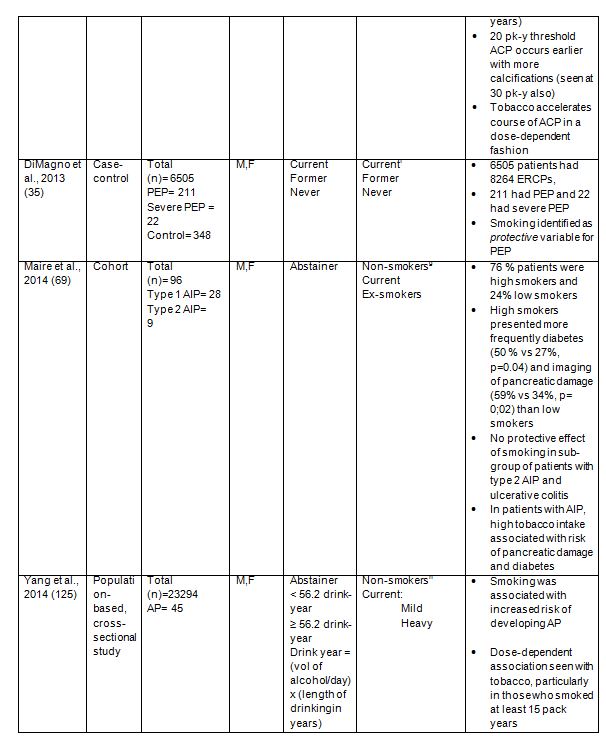
In addition to effects of tobacco smoking in development of AP and CP, the influence of smoking on the course of autoimmune pancreatitis (AIP) and post-endoscopic retrograde cholangiopancreatography (ERCP) pancreatitis (PEP) has been considered (35, 69). In the AIP study it was reported that high smokers (>10 pack years) presented more frequently diabetes (50% vs. 27%) and imaging pancreatic damages (59% vs. 34%) than low smokers (69). In addition there was a trend to observe more pancreatic exocrine insufficiency (41% vs. 29%). These data suggest that smoking could influence the natural course of AIP, similarly to that seen with alcoholic CP, although the association between smoking and relapse of AIP was not significant. Type 2 AIP is associated with inflammatory bowel disease, especially ulcerative colitis (UC), and smoking has been shown to have a protective effect in UC, although the exact mechanisms are not understood (15, 91). In this study, 22 patients had UC in association with AIP, although a protective effect of smoking on AIP course was not observed. This lack of effect could have been due to an irrelevant statistical analysis owing to small patient numbers. In the study evaluating tobacco effects on the course of PEP it was found that current smoking was independently protective against PEP (35). That current smoking is protective against PEP apparently contradicts the clinical observations outlined earlier in this review, that smoking is an independent, dose-dependent risk factor for AP and CP. However the protective effect observed in PEP may occur through nicotine, a major toxic component of tobacco (see section on Nicotine), which activates the nicotinic anti-inflammatory pathway, and can reduce pancreatic inflammation (48, 73, 109, 113). Nicotine can also relax the sphincter of Oddi in experimental models and might reduce sphincter spasm and obstruction which can cause PEP (12).
Although numerous clinical studies now substantiate an independent role for smoking in pancreatitis, it is often not acknowledged by physicians as a risk factor for the disease. In a study of 535 patients diagnosed with CP, 382 (71.4%) reported smoking, yet physicians recorded smoking as a risk factor for only 173 (45.3%). There was also a tendency to do so if the patient was a current smoker, reported elevated levels of smoking and/or had a concurrent alcohol problem (123, 124). The importance of smoking as an independent risk factor, particularly for chronic pancreatitis, is becoming vital for interventional purposes in light of new clinical information. One recent study used a questionnaire to i) investigate patient awareness regarding an association of smoking and pancreatic disease; ii) assess doctor-patient communication regarding smoking in general, and pancreatic disease specifically, and iii) examine the patient's stage of change for quitting smoking (47). Eighteen patients (mean age 52 years; 85% male) were used for the analysis. The data breakdown revealed that 56% of patients were aware of a connection between smoking and CP and 72% were conscious of alcohol and its role in pancreatitis. Patients conveyed that physicians were a critical reference source for their knowledge concerning causes of CP, although only 39% stated that their physician had directly referred to the effect tobacco has on the pancreas. This study highlights that efforts should be directed towards enhancing physician's knowledge on smoking and pancreatic disease as well as patient education.
In addition to continuing clinical studies, and relating newly-relevant information to patients, greater understanding is needed in defining which particular toxins in tobacco may initiate pancreatic disease at the cellular level. The fundamental biological mechanisms of tobacco-related pancreatitis remain largely uncharted and greater research is necessary so that potential therapeutic targets can be determined. An overview of current scientific findings relating to tobacco and pancreatitis are will follow
Scientific Evidence
In the following section, the effects of tobacco smoke on the pancreas will be explored and tobacco-specific toxins and their potential for generating pancreatitis through certain cellular pathways will be considered.
Cigarette Smoke
Of the 4000 chemicals in cigarette smoke, more than 60 have been recognized as prospective carcinogens. Tobacco smoke components, particularly nicotine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), and nitrosamines specific to tobacco, have been studied in cells and in vivo (2, 10, 20, 48, 89, 111, 117, 118). NNK is one of the most potent, as determined by studies in laboratory animals (90). N'-nitrosonornicotine (NNN) and Diethylnitrosamine (DEN) are two more nitrosamines derived from nicotine (95) that are potentially formed via nitrosation during processing of tobacco (49). Approximately 46% of NNN and 26%-37% of NNK in tobacco is preformed and the remainder is pyrosynthesized from nicotine during smoking (36). Other harmful constituents of tobacco smoke include polycyclic aromatic hydrocarbons (PAHs), although their role in pancreatic disease remains unclear (7, 36). In the following sections we will report the general effects of tobacco on the pancreas and subsequently focus on nicotine and NNK, since they are the most studied with respect to pancreatic disease.
Effects on Human Pancreas
In light of the medical evidence linking tobacco smoking and pancreatitis, closer attention has been paid to smoking-induced changes in pancreatic tissue from patients enrolled in such studies (53, 56, 60, 98, 99, 102, 112). Some of those changes are highlighted below.
Pancreatic Fibrosis: One study assessed pancreatic fibrosis (PF) in smokers versus non-smokers and found total PF and intralobular PF was significantly more present in smokers (total: 42.9 vs. 26.5%, P=0.027 and intralobular: 39.3 vs. 15.6%, P=0,013) (112). Since Pancreatic Stellate Cells (PSCs) are key players in PF (53, 97) it is highly likely that oxidative stress induced by tobacco components and cigarette smoke could lead to their activation, eventually resulting in PF.
Oxidative Stress: Expression of the pro-inflammatory cytokine interleukin-6 (IL-6) and antioxidants in pancreatic fluids and tissues in patients (both smoking and non-smoking) with CP have been measured (98). In comparison to non-smoking patients and healthy subjects, statistically higher levels of IL-6 and metallothionein, as well as increased activities of antioxidants (glutathione peroxidase; copper-zinc superoxide dismutase) were observed in smoking patients with CP. These observations further underscore the role oxidative stress may play in induction of tobacco-related pancreatitis.
Secretion: Several studies have assessed the effects of tobacco smoking on factors that affect both endocrine and exocrine pancreatic secretion in smokers versus non-smokers (56, 99). Numerous publications indicate there is decreased insulin secretion in both smoking patients and smokers with CP, and higher blood glucose levels were detected in the latter (59, 74, 119). These changes paralleled adaptations in pancreatic structure and altered endocrine function of the organ resulting from smoking (74). A further study looked at immunohistochemical localization of somatostatin and pancreatic polypeptide (two hormones that regulate secretion) in the pancreas of smoking and non-smoking patients with CP versus healthy controls (99). In pancreatic samples from smoking patients, significantly higher immunostaining of the hormones was detected, suggesting that tobacco smoking may partake in endocrine disturbances during CP development. Another retrospective study compared pancreatic duct cell function in smokers (current and past) with never-smokers by measuring secretin-stimulated peak bicarbonate concentration ([HCO3-]) in endoscopically-collected pancreatic fluid (56). Smoking (odds ratio, OR: 3.8, 95% CI: 1.6-9.1; P=0.003) and definite chronic pancreatitis imaging (OR: 5.7, 95% CI: 2.2-14.8; P<0.001) were determined to be independent predictors of low peak pancreatic fluid [HCO3-], controlling for age, gender, and alcohol intake. Furthermore, no interaction between smoking status and alcohol intake was observed in predicting duct cell dysfunction (P=0.571). Thus, measurement of pancreatic fluid bicarbonate in smokers reveals that cigarette smoking (past and current) is an independent risk factor for pancreatic duct cell secretory dysfunction (low pancreatic fluid [HCO3-]). Furthermore, the risk of duct cell dysfunction in subjects who smoked was approximately twice the risk (RR: 2.2) in never smokers.
Endothelin-1: Endothelin-1 (ET-1) is a protein that plays a role in blood vessel constriction and recent evidence has shown that it may be another marker of tobacco-linked pancreatitis (100, 101). Plasma ET-1 levels were nearly two-fold higher in smokers when compared to healthy controls. Histopathological analysis of pancreatic tissue demonstrated an increase in ET-1 levels in smokers and smokers suffering from CP. These findings may account for changes in blood flow to the pancreas seen during pancreatitis.
Pancreatic Dysfunction/protein Catabolism: One study examined levels of creatinine, uric acid and urea in non-smoking and smoking patients with CP (102). Their results show elevated creatinine and uric acid levels 1.5 times higher in the smoking group than that seen in the healthy controls. These findings would suggest that cigarette smoking may be an important factor in potential changes in uric acid levels in patients with CP. In addition, the decreased protein catabolism observed in this study is likely the result of progressing exocrine pancreatic dysfunction, seen in both smoking and non-smoking patients with CP.
Genetic Mutations: Chymotrypsinogen C (CTRC) is known to protect the pancreas by degrading the prematurely activated zymogen, trypsinogen. Rare mutations in CRTC prevent it from degrading trypsinogen and are associated with RAP and CP (128). The occurrence of such mutations in patients was evaluated from the North American Pancreatitis Study cohort II and it was found that a genetic variant, CTRC Variant G60G (c.180T), acted as a disease modifier, promoting progression of RAP to CP, particularly in the smoking population (60). The mechanism of how tobacco smoke or toxins interact with variants of CRTC to produce this disease phenotype is not yet clear.
Thus it seems the effects of tobacco exposure on human pancreas are numerous, and the physical and functional changes it produces are becoming more evident. However, precise cellular mechanisms and pathways which mediate these events are unclear. Identification of potential disease markers, some of which have been detected by assessment of pancreatic tissue from clinical studies, could prove useful in determining which cellular pathways to research and in designing appropriate experimental models for smoking–related pancreatitis. A crucial assumption is that by understanding the mechanism of disease, opportunities will arise for the development of therapeutic strategies.
Effects on pancreas in animal models of cigarette smoke exposure
Cigarette smoke consists of a complex mixture of compounds, so the development of dependable animal models of smoking and pancreatitis has been challenging. Specific compounds and mixtures which are most likely responsible for human disease have to be considered along with the route of administration (i.e. inhalational vs. systemic) and dosing to parallel the human experience. So far, only a small number of reasonable animal models of tobacco-related pancreatitis have been established (3, 20-27, 30-32, 48, 65, 118).
In one of the earliest animal models, rats were given ethanol intravenously under anesthesia and were exposed to cigarette smoke at 15 and 45 minutes (40 puffs; 2 minute session each time by mechanical ventilation) from the start of ethanol infusion. The investigators determined that this regimen would obtain nicotine plasma levels comparable to those found in human smokers (nicotine concentrations, 4–72 ng/mL; mean, 33 ng/mL). They found that cigarette smoke exacerbated pancreatic ischemia initiated by ethanol. In addition, cigarette smoke by itself elevated leukocyte-endothelium interactions and, in combination with ethanol, augmented pancreatic sequestration (48).
In another model of rat pancreatitis, tobacco smoke was administered through inhalation for 12 weeks. Animals that received high-dose exposure (160mg/m3) developed pancreatic damage consistent with that seen in CP and had boosted levels of the pancreatic zymogens, trypsinogen and chymotrypsinogen. These rats also developed focal pancreatic lesions with areas of increased extracellular matrix, although the pancreatic damage was reduced in comparison to that observed in human CP. These differences between the model and human CP might be due to the relatively short experimental time period (117). An additional report found that environmental tobacco smoke altered gene expression in the exocrine pancreas, by modifying the ratio of trypsinogen to its endogenous inhibitor (pancreas-specific trypsin inhibitor; PSTI). While trypsinogen was elevated in smoke-exposed animals, the expression of PSTI was not up-regulated. These modifications rendered smoke-exposed animals prone to pancreatitis (118).
That these models mimic the features of human pancreatitis is promising, but it does not yield much information as to which toxins are initiating pancreatitis and what their cellular targets may be. Other approaches have focused on specific toxins in tobacco, which may be likely candidates for initiating pancreatic disease. As mentioned earlier these include nicotine and the tobacco-specific nitrosamine, NNK. Their role in pancreatic and other cancers has been largely explored and will be considered later. In the sections that follow we will describe findings from animal models which explore effects of nicotine and NNK in development of pancreatitis.
Nicotine: Nicotine is a significant toxin in tobacco and cigarettes and may influence the development of pancreatitis and pancreatic cancer. Nicotine is rapidly absorbed in the lungs and is removed from the body within 120-180 minutes (28) . Nicotine metabolism occurs primarily through the cytochrome P450 (CYP) 2A6 pathway along with additional enzymes including aldehyde oxidase 1, UDP-glucuronosyltranferases, flavin-containing monooxygenase 3 and other CYPs e.g. 2A13, 2B6. Polymorphisms in CYP2A6 have been associated with racial and genetic differences in metabolism of nicotine, but it is unclear if these impact smoking-related pancreatic disease (77). Patients with CP and pancreatic cancer, when compared to healthy controls, have been found to have raised levels of P450 enzyme (27). Studies in which rats inhaled 3H-nicotine have shown that it accumulates in the pancreas and intestine (22, 27). In addition, in human pancreatic juice collected from smokers, elevated levels of nicotine metabolites have been measured. Cotinine, a primary metabolite, was present at levels around 130 ng/ml whereas NNK ranged from 1.37 ng/ml to 600 ng/ml (0.7µM and 6.6nM - 3µM respectively)(84).
Several studies have established the pathological and functional effects of nicotine on the pancreas. In a rodent model, rats were exposed to graded doses of nicotine either by aerosol, intragastric or ad-libitum feeding over a period of 3 to 16 weeks. Exocrine pancreatic cells from these animals exhibited cytoplasmic swelling, vacuolization, pyknotic nuclei and karyorrhexis. Furthermore, isolated acinar cells either treated with nicotine or harvested from nicotine-exposed animals showed similar cellular damage. These changes reflect those observed in acute or experimental pancreatitis (Figure 1) (20, 24, 25, 29, 30, 65). Nicotine also altered pancreatic secretion: it decreased pancreatic amylase secretion in rats, accompanied by retention of pancreatic zymogens (20, 21, 24, 25, 29-31). A subsequent study has shown that nicotine-induced secretory events in isolated rat acini are abrogated following treatment with mecamylamine, a nicotinic receptor antagonist, and some calcium channel antagonists (31). This pharmacologic evidence insinuates that nicotine modulates its responses through a nicotinic acetylcholine receptor (nAChR) and that calcium acts as a downstream effector. Nicotine also has been shown to change circulating levels of the gastrointestinal hormones gastrin and CCK in rats (26). Fluctuating basal levels of these hormones, and serum enzymes such as amylase and lipase, have been related to morphological variations in pancreatitis (21, 27). Nicotine can also regulate lipid peroxidation and oxidative stress although it is undetermined if these processes take part in the pathophysiology of pancreatitis (21).
Nicotine exposure may lead to increased expression of proteins that contribute to pancreatitis and other pancreatic diseases. One study used mass spectrometry-based proteomics to investigate the effects of nicotine on the proteomes of two pancreatic duct cell lines–an immortalized normal cell line (HPNE) and a cancer cell line (PanC1) (79). With more than 5,000 proteins identified per cell line, over 900 were differentially expressed following nicotine treatment, and 57 of these proteins were found in both cell lines. In a prior study, nicotine treatment had been shown to increase expression of amyloid precursor protein (APP) in pancreatic stellate cells (80) and in this later study, up-regulation of APP was also observed in both ductal cell lines. Thus nicotine-mediated expression of APP might be linked with inflammatory or fibrotic responses in pancreatitis.
NNK: NNK, a tobacco-specific nitrosamine derived from nicotine, is one of the most toxic components in cigarette smoke. The role of NNK as an initiator of, and sensitizer to, AP was revealed in studies using isolated rat acinar cells and in vivo models of pancreatitis (4). Firstly, NNK was found to induce a key event in initiation of pancreatitis: premature activation of digestive zymogens (trypsinogen and chymotrypsinogen).
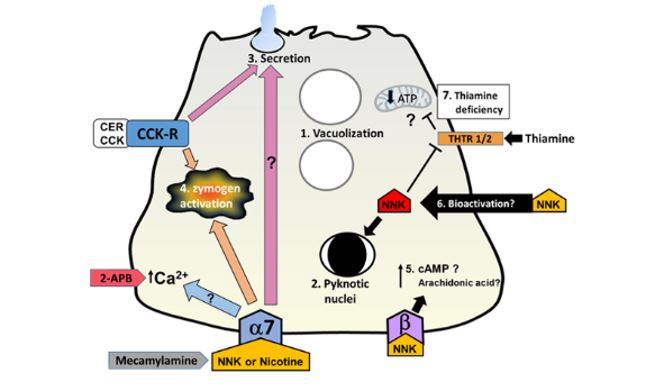
Figure 1. Cellular mechanisms mediated by nicotine and NNK in pancreatic acinar cells. Exposure to nicotine and NNK is known to cause morphological changes comparable to those seen in pancreatitis including 1. vacuolization and 2. pyknotic nuclei. 3. Secretion: nicotine stimulates secretion by itself and augments cholecystokinin-mediated (CCK) secretion at low concentrations (100 uM); at higher concentrations (>1 mM) it inhibits secretion. Pre-treatment of pancreatic acinar cells with the nAChR blocker mecamylamine reduces nicotine-mediated effects. The calcium channel antagonist 2-APB also prevents nicotine-stimulated events; this implies that nicotine-sensitive pathways involve the alpha-7 nAChR and intracellular calcium signals. 4. Zymogen activation: NNK induces zymogen activation in acini and augments cerulein (CER)-induced zymogen activation; this effect is abrogated by mecamylamine and in α7-/- mice. 5. Elevations in cAMP and arachidonic acid through Beta-adrenergic receptor signaling: NNK binds to beta-adrenergic receptors with high affinity. Pre-incubation of acini with beta blocker propranolol did not block NNK-mediated zymogen activation, therefore this pathway does not mediate this process. Whether NNK elevates levels of the second messengers cAMP and arachidonic acid, causing other pancreatitis responses is undetermined. 6. Bioactivation: NNK can be taken up by cells and converted to bioactive forms by cytochrome P450 enzymes; this occurs in the pancreas but it has not been determined if these bioactive forms participate in pancreatitis. Bioactivated NNK can affect cell function at the transcriptional level. 7. Thiamine deficiency and mitochondrial dysfunction: NNK has been shown to inhibit uptake of the vitamin thiamin, by reducing levels of thiamin transporters; whether this is via a bioactivated form of NNK is unclear. Thiamin is crucial for pancreatic function due to its role in metabolism and as a cofactor for multiple enzymes in mitochondrial ATP production. Thiamin deficiency may decrease cellular ATP levels, leaving the pancreas vulnerable to a secondary insult and thus development of pancreatitis.
Secondly, the effects of NNK in conjunction with a commonly-used model of pancreatitis were determined, to see if NNK pre-treatment could augment pancreatitis responses. The model of experimental pancreatitis employed was the ‘cerulein’ model. Cerulein is an orthologue of the hormone cholecystokinin (CCK) and, when given at supraphysiologic concentrations (10-100x that required to induce physiological responses), it causes typical pancreatitis responses (zymogen activation, histological/morphological changes) in isolated acinar cells or live animals. NNK pre-treatment in the cerulein model elevated zymogen activation above that seen with NNK or cerulein treatment alone. Furthermore, NNK triggered cellular injury in the pancreas (vacuolization, pyknotic nuclei, and edema) similar to that typically seen during AP. These findings raise the question: how does NNK mediate these pancreatitis responses?
NNK is a high-affinity agonist of β-adrenergic receptors and nicotinic acetylcholine receptors (nAChR), particularly the α7 isoform, and could influence the development and progression of pancreatic diseases through these receptor-mediated pathways. NNK is structurally similar to classic β-adrenergic agonists and has been found to bind with high affinity to human β-1 and β-2 receptors (EC50 for β1= 5.8 nM; EC50 for β2 = 128 nM) (96). In mammalian cells, activation of β-adrenergic receptors triggers adenylate cyclase to generate the second messenger cAMP, although in some cells it can cause release of arachidonic acid. Elevations in cAMP may participate in pancreatitis responses (19). Although one study detected β- adrenergic receptors in rat acinar cells, NNK mediated zymogen activation was not abrogated when β-adrenergic receptors were inhibited with propranolol (3). It is possible that NNK mediates arachidonic acid release through phospholipase A2 (PLA2), an important factor in inflammation. Various isoforms, namely phospholipase A2-II and A2-IV, are elevated during human AP and may affect both local disease severity and systemic complications (43). Whether NNK mediates other pancreatitis responses through these receptors is undetermined.
More recently, attention has focused on the possibility of NNK initiating pancreatitis responses through a non-neuronal form of the α7 isoform of nAChR. These receptors were initially described within the nervous system, but have subsequently been identified in non-neuronal cells (95). Various cancer cell lines, human keratinocytes, and epithelial cells all have α7 nAChR and respond to NNK exposure (EC50 for NNK= 0.03 µM). NNK is present in tobacco smoke at concentrations 5000-10000 times less than nicotine yet it exhibits 1000-fold higher affinity for α7 nAChR by comparison. In addition, up-regulation of α7 nAchRs are observed in the organs of smokers, and experimentally in the pancreas and lungs of rodents following chronic exposure to nicotine or NNK (2, 95).
In order to determine whether NNK mediated pancreatitis through a non-neuronal α7 nAChR, it was first established that the receptor was present in rat pancreatic acini through PCR analysis (4). Next, a functional role was revealed when isolated acini were pre-treated with the nAChR antagonist mecamylamine, and NNK induced zymogen activation was abrogated. These findings were further validated by studies using transgenic mice. NNK treatment in acini isolated from α7 nAChR-/- mice failed to elicit zymogen activation when compared with wild type (9).
NNK may mediate pancreatitis responses through a direct interaction with α7 nAChR on the acinar cell surface, but it might also wield influence over inflammatory cells during pancreatitis (Figure 2). α7nAChRs are expressed on macrophages, and both NNK and nicotine could potentially modulate immune responses. Nicotine hinders generation of pro-inflammatory cytokines from macrophages by inhibiting the NFκB pathway, which mediates macrophage activation (25, 115). Furthermore, in one study, the treatment of mice with mecamylamine (the general nAChR blocker) decreased neutrophil and macrophage migration to pancreatic tissue and led to more severe experimental pancreatitis (113). In a subsequent investigation, prophylactic and delayed therapeutic application of nicotine significantly attenuated the severity of acute experimental pancreatitis in rats through stimulation of the cholinergic anti-inflammatory pathway (94). Nicotine pre-treatment had a protective effect in a model of severe acute pancreatitis (SAP), in which mice were given a retrograde injection of 2% Na-taurocholate into the pancreatic duct (127). Nicotine (50-300 µg/kg) reduced tissue injury, enzyme production, and generation of pro-inflammatory cytokines. Nicotine also up-regulated CD4+ CD25+ regulatory T cells (Treg) through increased expression of immunoregulatory molecules and secretion of transforming growth factor β1 (TGF-β1).
The notion of nicotine and NNK inducing an anti-inflammatory response and reducing pancreatitis severity may seem contrary to other studies showing tobacco toxins promoting the disease (4). However, it has also been shown that prolonged exposure to cigarette smoke results in chronic inflammation in the pancreas. Other studies have indicated that NNK may actually initiate pro-inflammatory effects in macrophages and other cells through its uptake and metabolism. In U937 human macrophages NNK gets absorbed and metabolized, undergoing a process known as ‘bioactivation’ (89). Bioactivation of NNK happens via the cytochrome P450 (CYP450) enzyme family through three primary pathways: (a) carbonyl reduction, (b) pyridine N-oxidation and (c) alpha-hydroxylation. Following bioactivation, NNK metabolites subsequently activate NFκB, inducing release of TNF-α while inhibiting synthesis of interleukin-10 (IL-10). Decreased levels of other cytokines and modulators namely interleukins (IL-2, IL-6), granulocyte/macrophage-colony-stimulating factor (GM-CSF) and macrophage chemotactic protein 1 (MCP-1) are also seen (89).
Thus it seems that the effects of NNK and nicotine in pancreatitis are multi-faceted and might appear ambiguous. Early pancreatitis events may consist of a combination of direct interaction of NNK/nicotine with α7 nAChR on acini and a possible anti-inflammatory phase through α7 nAChR localized on macrophages (4, 94, 113, 115).

Figure 2. Inflammatory events in smoking-related pancreatitis. Early events in smoking-related pancreatitis include 1. zymogen activation through a direct interaction of NNK with α7 nAChR on pancreatic acini and 2. stimulation of the cholinergic anti-inflammatory pathway by NNK/nicotine binding to α7 nAChR localized on macrophages. The anti-inflammatory phase could be an initial response that ultimately yields to a chronic inflammatory phase with continued exposure to cigarette toxins. 3. Bioactivation: NNK gets taken up and metabolized by macrophages. 4. Pro-inflammatory response: the “bioactivated” derivatives of NNK can affect gene transcription and lead to activation of pro-inflammatory pathways. Activated macrophages invade damaged pancreatic tissue, giving rise to pancreatic inflammation.
The anti-inflammatory phase could be an initial response that ultimately yields to a chronic inflammatory phase with continued exposure to cigarette toxins (46). Chronic inflammatory responses happen much later, perhaps through uptake and metabolism (bioactivation) of NNK/nicotine in macrophages (Figure 2).
It is unclear whether bioactivation of NNK occurs in pancreatic acinar cells and contributes to tobacco-related pancreatitis. Although P450 enzymes, which are crucial for bioactivation of NNK, have been identified in rodents (isoforms 2B6, 3A5 and 2A3), there have been inconsistent results in human pancreas (1). One study using cytochemical detection methods, found no evidence of the P450 enzymes in human pancreatic samples from smokers and non-smokers (7). However, another study detected CYP450 enzymes in human pancreatic tissue using immunohistochemical methods (41). Furthermore, the levels of enzymes were elevated in the samples from patients with CP and pancreatic cancer (41). Thus metabolism of NNK within pancreatic cells may actually be a factor in development of smoking-related pancreatitis and other pancreatic diseases.
More recent findings have shown that NNK may effect changes within the pancreatic acinar cell at the genetic level (105). Whether this is through a “bioactivated” form of NNK or some other pathway remains unclear. The vitamin thiamin is critical for both exocrine and endocrine functions of the pancreas and pancreatic cells are known to maintain high levels via uptake from their surroundings. Uptake is achieved through thiamin transporters-1 and -2 (THTR-1 and THTR-2). Protein and mRNA levels of these transporters were significantly reduced when pancreatic acinar 266-6 cells were treated with NNK. These changes were further coupled with a decrease in thiamin uptake and thiamin transporter promoters- SLC19A2 and SLC19A3. Long term treatment of NNK in mice yielded similar results (105). This study highlights that cigarette toxins can cause alterations in pancreatic cells at a genetic level resulting, in this particular case, thiamin deficiency. Thiamin deficiency, followed by a drop in cellular ATP levels, might sensitize the pancreas to a secondary insult, predisposing to pancreatitis.
It is apparent, from all of the scientific studies described here, that tobacco smoke, in particular toxins such as nicotine and NNK, are capable of inducing diverse pancreatitis responses via multiple cellular mechanisms. It is likely, that through similar mechanisms, progression of chronic disease to cancer may occur. In the following sections, we will explore the clinical evidence between smoking and development of pancreatic cancers, and describe potential cellular mechanisms.
3. Role of Tobacco in Developmenet of Pancreatic Disease: Pancreatic Adenocarcinoma
Although there is much debate about the effect of alcohol abuse in causing and/or promoting pancreatic cancer, tobacco smoking is a major established risk factor for the disease (66, 84). Strong evidence indicates that cigarette smoking increases the risk of pancreatic cancer and accelerates the process of its development. Smoking increases the risk of pancreatic cancer up to 6-fold depending on the duration and intensity of smoking (52, 86, 116) and nearly one quarter of all pancreatic cancer deaths are linked to tobacco use (68). At least two different studies published recently showed that smokers are diagnosed with pancreatic cancer at ages 6 to 15 years younger than non-smokers (8, 70). Therefore, understanding the mechanisms through which smoking predisposes to pancreatic cancer is urgently needed. This will help target patients at high risk for the disease with preventive strategies, and permit development of treatment approaches directed at cell signaling pathways involved in smoking-induced pancreatic cancer.
In the next sections of this manuscript a short review of the clinical evidence is provided for the association between smoking and pancreatic cancer, and a more detailed review of the pathways mediating the pro-cancer effects of cigarette smoke compounds in the pancreas.
Clinical Evidence
Research on the association between smoking and pancreatic cancer goes back to the mid-sixties. One of the first studies to look at the association was published in 1970 where the author compared the age-adjusted death rates in the years 1964-1965 from cancers of different sites and the annual consumption of cigarettes in data collected from 20 countries. The author found no significant correlation between cigarette smoking and death from pancreatic cancer; although the risk of death from pancreatic cancer in smokers was non-significantly increased by a relative ratio of 1.21 in males and 1.15 in females (106). One possible reason for this result is that the comparison was performed between heavy smokers (smoking a number of cigarettes above the average of cigarettes smoked by the whole population analyzed, which is 3.5 cigarette per day) and light smokers (smoking a number of cigarettes below the average). Results from the same study showed nonsignificant increase in deaths from lung cancers in heavy smokers compared to light smokers confirming that the significance is lost because the threshold between light and heavy smokers was very low at 3.5 cigarettes per day. In 1973, the first study to show association between cigarette smoking and pancreatic cancer in humans was published by Wynder et al (121). Two years later, the same author showed an increased risk for pancreatic cancer associated with the number of cigarettes smoked per day. He found that smoking 1 to 10 cigarettes per day significantly increases the relative risk of pancreatic cancer by 2-fold, while 21-40 cigarettes per day increases it by 3.5-fold and smoking over 41 cigarettes per day increases the risk by 5-fold (120). Another group analyzing 38 case-control studies concluded that relative risk for pancreatic cancer among smokers is 2 to 4 times making it the third highest smoking-related cancer after the lung and the upper aero-digestive tract cancers (93). Depending on the duration and intensity of cigarette smoking, it could increase the risk of pancreatic cancer up to 6-fold (52, 85, 116). In addition to these findings, several other studies have assessed the association between smoking and pancreatic cancer and have emphasized cigarette smoking as a major risk factor for the disease(66, 85).
Nearly one quarter of all pancreatic cancer deaths are linked to tobacco use (33). Furthermore, two different studies published recently highlighted that diagnosis of pancreatic cancer occurs 6 to 15 years earlier in smokers when compared with non-smokers (8, 70). Compared to non-smoking, smoking less than one pack a day decreased the age of diagnosis by 3 years, and 6 years for those smoking more than two packs of cigarettes per day (8).
However, the deleterious effect of tobacco smoking is not perpetual. Indeed, a study in 1986 showed a strong association between smoking and increased risk for pancreatic cancer, but the effect disappeared after a decade of non-smoking (68). The deleterious effect of smoking did not change in smokers who stopped smoking less than 10 years as the median age of diagnosis was similar to smokers; whereas, it did completely resolve in patients who stopped smoking for more than 10 years as the age of diagnosis was similar to non-smokers (8). The same study showed that the proportional hazard ratio among smokers was similar to those who stopped smoking less than 10 years ago (relative ratio 1.65 and 1.27, respectively) compared to a relative risk of 0.95 for those who stopped smoking more than 10 years ago (8).
It is worth noting that studies in animal models showed that exposing wild type mice to cigarette smoke compounds may cause pancreatic lesions, but it rarely reaches the pancreatic adenocarcinoma stage suggesting that smoking cooperates with other environmental and/or genetic factors such Kras mutation to induce pancreatic cancer. In fact, consuming NNK and NNAL (0.5, 1 and 5ppm) in drinking water for a lifetime by rats did not induce pancreatic ductal adenocarcinoma, except in 13% of rats treated with the high dose of 5ppm of NNAL (90).
Scientific Evidence
Smoking and Genetic Mutations
Analysis of known pancreatic cancer mutations in the pancreatic tissue of patients show a significant increase in the number of mutations per tumor in smokers (53.1 mutations per tumor) compared to 38.5 mutations per tumor in never smokers (16).
Kras mutation is a major mutation associated with pancreatic cancer and is present in more than 90% of pancreatic cancer patients (5). A strong association exists between Kras mutation and smoking in lung cancer patients; however such a link is hard to establish in pancreatic cancer. The meta-analysis study performed by Porta et. al showed no-significant association between Kras mutations and smoking status in pancreatic and colorectal cancers whereas significant association was found in lung cancer (82). The sequencing of the pancreatic cancer genome found that the difference in the total number of mutations between smokers and non-smokers was not driven by mutations of the known driver genes in pancreatic cancer such as Kras, p53, p16/CDKN2A and SMAD4, but instead was predominantly observed in genes mutated at lower frequency (16). In addition, no differences were observed in mutations in carcinomas from the head versus tail of the gland. The same study by Porta et. al revealed a very important observation related to the spectrum of mutations of Glycine 12 in the Kras protein in the 3 cancers. In pancreatic and colon tumors, 85% and 74% of the mutations are Val or Asp in pancreatic and colon cancers, respectively, whereas in lung cancer only 37% are Val or Asp against 49% Cys compared to 3% and 8% Cys in pancreatic and colon cancers, respectively (82). This data suggest a possible association between the spectrum of mutations and the effect of smoking.
The difference between the spectrum of Kras mutations between pancreatic and lung cancer suggests using it to differentiate between metastatic pancreatic tumors found in the lung and the primary lung tumors. This is extremely important knowing that the survival of patients between the two cases is significantly different and the treatment to be applied is therefore altered. Analysis of the Kras mutations revealed that the presence of the KRAS G12C mutation had 96% specificity and positive predictive value for lung adenocarcinoma, whereas G12R was 99% specific for pancreatic cancer with a positive predictive value of 86% (57). However, it is worth nothing that although Kras mutation is not significantly induced by smoking in the pancreas, recent data indicate that nicotine further stimulates mutated Kras activation leading to more aggressive pancreatic tumors in animal models of the disease (50). The cell signaling mechanism mediating this effect will be discussed in the subsequent section of this review. Other less common mutations associated with pancreatic cancer include p53, BRaf, Mek and Cox2, and deletion of SMAD4 and p16INK4A (44).
Analysis of the DNA adducts induced in rat lung and pancreas after treatment with different doses of NNK or NNAL showed that both compounds induced similar DNA adducts in both organs. However, the level of DNA adducts was significantly lower in the pancreas compared to the lung explaining, at least partially, why a higher level of DNA mutations is observed in the lung of humans and animals exposed to cigarette smoke compared to the pancreas (13).
Smoking Compounds and Cell Signaling in the Pre-Cancer and Cancer Cells
As mentioned before, cigarette smoke contains over 4000 chemicals with at least 60 of them being carcinogens. Of these constituents, nicotine and NNK, in addition to cigarette smoke extracts, are the most studied in the context of cancer and will be reviewed in the following sub-sections.
Cell Signaling Pathways Affected By Nicotine
As referred to earlier in this article, mass spectrometry-based proteomics analysis of pancreatic ductal and cancer cell lines exposed to nicotine showed that over 900 proteins were significantly abundant upon nicotine treatment, 57 of which were found in both cell lines. However, most of the proteins regulated by nicotine were different between the two cell types suggesting that nicotine may play different roles in the initiation and progression of pancreatic cancer (79).
Nicotine was shown to stimulate cancer promotion in several animal models of pancreatic cancer. Administration of nicotine accelerated transformation of pancreatic cells and tumor formation in the elastase-Kras (Ela-Kras) and the KrasLSL-G12D/+; Trp53LSL-R172H/+; Pdxcre/+ (KPC) mice. Nicotine induced dedifferentiation of acinar cells by activating the Akt-Erk-Myc signaling; this leads to the inhibition of Gata6 promoter activity, loss of GATA6 protein, and subsequent loss of acinar differentiation and hyper-activation of oncogenic Kras (50).
Sustained exposure to nicotine induced activation of Akt and Erk kinases, both important pro-cancer pathways (14). Nicotine treatment in the drinking water for 4 weeks significantly reduced the therapeutic response of mouse xenografts to gemcitabine. This was associated with decreased gemcitabine-induced caspase-3 cleavage and inhibition of phosphorylated/ activated forms of Akt, Erk and Src in xenograft tissues (14).
Nicotine also promoted aggressiveness of established tumors as well as the epithelial–mesenchymal transition, increasing numbers of circulating cancer cells and their dissemination to the liver, compared with mice not exposed to nicotine. Nicotine induced pancreatic cells to acquire gene expression patterns and functional characteristics of cancer stem cells. These effects were markedly attenuated in KPC mice given metformin, which prevented nicotine-induced pancreatic carcinogenesis and tumor growth by up-regulating GATA6 and promoting differentiation toward an acinar cell program (50). Gata6 ablation renders acinar cells more sensitive to the Kras mutation in a mouse model of pancreatic cancer thereby accelerating tumor development. Furthermore, Gata6 expression is spontaneously lost in a Kras mouse model of pancreatic cancer in association with altered cell differentiation (72). Gata6 is a transcription factor that plays a tumor-suppressor role through the promotion of cell differentiation, the suppression of inflammatory pathways, and the direct repression of cancer-related pathways. Epidermal growth factor receptor (EGFR) pathway is an example of the pro-cancer pathways inhibited by GATA6 as its activity is up-regulated in the normal and pre-neoplastic Gata6-null pancreas (72).
In a different study Trevino et al. showed that stimulation of pancreatic cancer cells with nicotine concentrations that are within the range of human exposure results in activation of Src kinase and the induction of the inhibitor of differentiation-1 (Id1) transcription factor. Depletion of α7-nAChR or Id1 prevented nicotine-mediated induction of proliferation and invasion of pancreatic cancer cells in vitro (34, 110). In addition, they found that nicotine confers resistance to gemcitabine in pancreatic cancer cells and that Src or Id1 depletion prevents the nicotine-induced resistance. Their data show that nicotine promotes the growth and metastasis of pancreatic cancer, and confers resistance to gemcitabine in vivo in an orthotopic model of pancreatic cancer (110). Of note, Src kinase plays a major role in promoting pancreatic cancer as it has been shown that it regulates cancer cell proliferation, invasion, and metastasis (54, 67). Clinical analyses of resected pancreatic cancer specimens demonstrated a statistically significant correlation between phospho-Src, tumor grade/differentiation, and worsening overall patient survival (110). Other pathways involved in the pro-cancer effect of nicotine include the STAT3/MUC4 pathway (75). This will be discussed in more detail in the next section.
Figure 3 is a representation of important pathways demonstrated to mediate the pro-cancer effects of nicotine in the pancreas.
Cell Signaling Pathways Affected by NNK and Cigarette Smoke Extract
Similarly to the effect of nicotine on the Akt pathway, up-regulation of the Akt kinase phosphorylation/activation was shown to be induced by NNK and cigarette smoke extracts (CSE) in pancreatic ductal cells. This effect led to the inhibition of apoptosis and was mediated by NADPH oxidase (78). Long exposure to NNK and CSE led to the inhibition of autophagy as well (78). NNK also stimulated proliferation of the pancreatic ductal cells through a mechanism that involves activation of EGFR (11). Both studies demonstrate involvement of the EGFR/Akt pathway in the regulation of proliferation and resistance to apoptosis of the pancreatic ductal cells.
Cigarette smoke extract and its major component nicotine, significantly up-regulates MUC4 in pancreatic cancer cells. MUC-4 plays several roles in the progression of cancer, especially through its signaling and anti-adhesive properties which contribute to tumor development and metastasis. The smoking-induced MUC4 over-expression was via the α7-nAChR stimulation and subsequent activation of the JAK2/STAT3 downstream signaling cascade in cooperation with the MEK/ERK1/2 pathway (75). The MUC4 up-regulation promotes pancreatic cancer cell migration and Src kinase is involved in mediating this pro-metastasis effect. In vivo, cigarette smoke exposure significantly stimulated tumor metastasis to various distant organs in the orthotopic model of pancreatic cancer (75).
The effect of cigarette smoke on promoting pancreatic cancer is observed in the early pre-cancer stages of the disease. Indeed, exposure of the Pdx1-Cre;LSL-Kras (KC) mice to cigarette smoke for 20 weeks significantly accelerated the development of the pancreatic intraepithelial neoplasia (PanIN) lesions, the precursors of pancreatic adenocarcinoma. This effect was associated with stimulation of inflammation markers such as IFN-γ and CXCL2 as well as enhanced activation of pancreatic stellate cells (58).
Data from Edderkaoui et al. show similar up-regulation of PanIN lesion formation, inflammation, fibrosis and stellate cells activation in the same animal model, but with a shorter exposure to cigarette smoke (6 weeks) (38). Their data further demonstrated a significant stimulation of EMT in the PanIN cells in mice exposed to cigarette smoke suggesting that smoking may cause early metastasis of the pre-cancer PanIN cells before even reaching the cancer stage. Of note, EMT of the pre-cancer cells was observed in the KPC mouse model of pancreatic cancer. EMT was associated with expression of cancer stem cell properties and it was associated with dissemination of these cells to the liver; a phenomenon that preceded tumor formation in the pancreas (88). Very importantly, EMT of the cancer cells was also associated with abundant inflammatory response and treatment with immunosuppressive agents prevented dissemination of the pre-cancer cells (88).
Figure 3 is a representation of important pathways demonstrated to mediate the pro-cancer effects of NNK and cigarette smoke in the pancreas.
Smoking and Inflammation
Pancreatic cancer is characterized by a strong desmoplasia. Pro-inflammatory mediators have been associated with pancreatic diseases including pancreatitis and pancreatic cancer.
cancer. 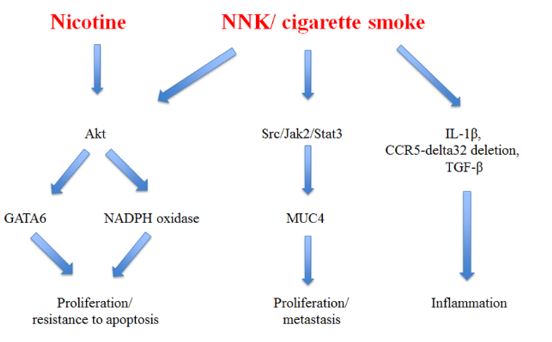
Figure 3: Pathways regulated by smoking compounds in pancreatic cells. Nicotine stimulates Akt kinases leading to activation of the NADPH oxidase and GATA6 pathways, therefore, stimulating proliferation and inhibiting apoptosis. NNK and cigarette smoke stimulate the same Akt pathways in addition to the Src/Jak2/Stat3 pathway leading to proliferation and metastasis as well as a pro-inflammation pathway.
Inflammatory cells as well as cytokines, chemokines, and their receptors have different biological functions including inflammatory response, angiogenesis, and metastasis. Strong evidence indicates that pro-inflammatory cytokines, chemokines, and their receptors are expressed in pancreatic cells and infiltrating immune cells within inflamed pancreatic tissues (45).
One major mechanism through which smoking induces inflammation of the pancreas, which may lead to pancreatic cancer, is through induction of pancreatitis. Mechanisms of smoking-induced pancreatitis were discussed earlier in this review. The next section will discuss the smoking-induced inflammatory pathways independently of pancreatitis.
It is well established that exposure to cigarette smoke stimulates inflammatory cell infiltration. In KC mice, cigarette smoke induced concurrent increase in the macrophage and dendritic cell (DCs) (58). NNK treatment significantly increases macrophage infiltration and expression of pro-inflammatory mediators such as macrophage inflammatory protein 1 alpha (MIP-1α, interleukin 1 beta (IL-1β), and TGF-β in mice neoplastic lesions (117). Higher infiltration of inflammatory cells including macrophage and mast cells is associated with strong expression of vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (b-FGF) in human pancreatic cancer tissues compared to normal pancreatic tissues (40).
Cytokines and other pro-inflammatory mediators have been implicated in inflammatory pancreatic diseases including pancreatitis and cancer. Analysis of cytokine gene polymorphisms as risk factors for pancreatic cancer suggested the possibility of interactions between current active smoking and the CCR5-delta32 deletion allele. The age adjusted interaction ratio (95% confidence intervals) for CCR5-delta32 and smoking was 1.4 (37). Of note, macrophages highly express the chemokine (C-C motif) receptor (5CCR5), the receptor of thechemokine C-C motif ligand 5 (CCL5). CCL5 is an anti-tumor chemokine as it induces immunecell recruitment (45, 76). The data suggests that intact CCR5 may protect from smoking-induced pancreatic cancer (37).
Exposure of the Elastase-IL-1beta transgenic mice (a model of chronic pancreatitis) to aqueous CSE for up to 15 months induced a significant flattening of the pancreatic ductal epithelial cells and severe glandular atrophy compared with untreated transgenic mice. Ductal epithelial cells displayed a high proliferative index, minimal apoptosis, and induction of COX-2, all markers associated with pancreatic cancer (104). Of note, Cox2 can induce activation of oncogenic Kras leading to pancreatic inflammation and fibrosis, and development of pancreatic intraepithelial neoplasia lesions and pancreatic ductal adenocarcinoma (81).
Another important pathway through which smoking promotes pancreatic cancer is by prompting oxidative stress. Indeed, Nicotine induces oxidative stress in rat pancreas and this was associated with inflammation and increased IL-6 secretion in the pancreas (55). Of note, expression of inducible nitric oxide synthase (iNOS) is increased during the development and progression of pancreatic cancer as well as in inflamed tissues (42, 114).(42, 114)(43, 115)(43, 115)(42, 114)(42, 114)(42, 114)(42, 114)(42, 113)(42, 113)(42, 113)(42, 113)(42, 113)(42, 113)(42, 113)(42, 113)(42, 113)(42, 113)(42, 113)(42, 113)(42, 113)(42, 113)
Lastly, a study by Lazar et al. showed that cigarette smoking and nicotine may contribute to pancreatic cancer inflammation by inducing monocyte chemo-attractant protein-1 (MCP-1) and provide a novel insight into a unique role for osteopontin (OPN) in mediating these effects (62).
Smoking and Fibrosis
Published studies indicate that exposure of mice to cigarette smoke activates pancreatic stellate cells (SC) as expressed by the level of its marker alpha-smooth muscle actin (α-SMA) and induces expression of extracellular matrix proteins (38, 58).
In vitro, nicotine at levels in smokers' blood induced proliferation and up-regulated expression of collagen1-α2 and TGF-β1 in hepatic SC. This pro-fibrogenic effect of nicotine was through actions on nAChRs expressed on hepatic SC. Nicotinic receptor antagonists reversed the nicotine-induced pro-fibrogenic effects (103). Very little data is published on the effect of smoking compounds on pancreatic SC in vitro. A unique study published in 2014 showed that smoking compounds significantly increase pancreatic SC proliferation and migration. The study showed that pancreatic SC express the nAChR isoforms alpha 3, 5, 7 and epsilon (122).
It is anticipated that smoking compounds stimulate activation and proliferation of pancreatic SC and promote deposition of extracellular matrix proteins by SC. This would result in a micro-environment favorable for proliferation of cancer cells and resistance to apoptosis.
Mouse, rat, and human pancreatic SC have slightly different morphologies in their untreated states. Following nicotine treatment, pancreatic SC acquire a slightly different morphology compared to the untreated cells and are characterized by a more elongated shape caused by narrow cytoplasmic projections.
Mass spectrometry analysis of pancreatic stellate cells exposed to nicotine show that of the total proteins identified 25-30% were exclusive to either nicotine-treated or untreated cells. Such nicotine-induced proteins included collagen alpha1(III) chain and alpha1(V) chain (80).
4. Conclusions
For over four decades studies have demonstrated that cigarette smoking is a major risk factor for pancreatic cancer. More recently, smoking was found to potentiate alcohol-induced pancreatitis. However, in the last decade, strong evidence confirms an independent effect of smoking in promotion of acute and chronic pancreatitis. However, despite the vast amount of data confirming an association between smoking and pancreatic diseases, data exploring the cellular mechanisms involved are less accepted. Various well known pro-pancreatitis pathways (e.g. NF-kB) and pro-cancer pathways (e.g. Akt kinase) are shown to be up-regulated in pancreatic cells. More comprehensive studies need to be performed to determine the detailed mechanism of interaction between these pathways and the receptors stimulated by cigarette smoke compounds. Furthermore, the effect of smoking on cells recruited and activated in the pancreatic tumor microenvironment and during acute and chronic pancreatitis requires further investigation.
5. References
- Akopyan G and Bonavida B. Understanding tobacco smoke carcinogen NNK and lung tumorigenesis. Int J Oncol 29(4): 745-752, 2006. PMID: 16964372.
- Al-Wadei HA and Schuller HM. Nicotinic receptor-associated modulation of stimulatory and inhibitory neurotransmitters in NNK-induced adenocarcinoma of the lungs and pancreas. J Pathol 218(4): 437-445, 2009. PMID: 19274673.
- Alexandre M, Pandol SJ, Gorelick FS and Thrower EC. The emerging role of smoking in the development of pancreatitis. Pancreatology 11(5): 469-474, 2011. PMID: 21986098.
- Alexandre M, Uduman AK, Minervini S, Raoof A, Shugrue CA, Akinbiyi EO, et al. Tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone initiates and enhances pancreatitis responses. Am J Physiol Gastrointest Liver Physiol 303(6): G696-704, 2012. PMID: 22837343.
- Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N and Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 53(4): 549-554, 1988. PMID: 2453289.
- Alsamarrai A, Das SL, Windsor JA and Petrov MS. Factors that affect risk for pancreatic disease in the general population: a systematic review and meta-analysis of prospective cohort studies. Clin Gastroenterol Hepatol 12(10): 1635-1644, 2014. PMID: 24509242.
- Anderson KE, Hammons GJ, Kadlubar FF, Potter JD, Kaderlik KR, Ilett KF, et al. Metabolic activation of aromatic amines by human pancreas. Carcinogenesis 18(5): 1085-1092, 1997. PMID: 9163700.
- Anderson MA, Zolotarevsky E, Cooper KL, Sherman S, Shats O, Whitcomb DC, et al. Alcohol and tobacco lower the age of presentation in sporadic pancreatic cancer in a dose-dependent manner: a multicenter study. Am J Gastroenterol 107(11): 1730-1739, 2012. PMID: 22929760.
- Ashat M, Tashkandi N, Sreekumar B, Patel V, Chowdhury AB, Shugrue C, et al. Sa1788 Tobacco Toxin NNK (4-[Methylnitrosamino]-1-[3-Pyridyl]-1-Butanone) Mediates Zymogen Activation in Murine and Human Pancreatic Acini. Gastroenterology 146(5): S-296. (Abstract) 2014.
- Askari MD, Tsao MS and Schuller HM. The tobacco-specific carcinogen, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone stimulates proliferation of immortalized human pancreatic duct epithelia through beta-adrenergic transactivation of EGF receptors. J Cancer Res Clin Oncol 131(10): 639-648, 2005. PMID: 16091975.
- Bagcivan I, Kaya T, Turan M, Goktas S, Demirel Y and Gursoy S. Investigation of the mechanism of nicotine-induced relaxation on the sheep sphincter of Oddi. Can J Physiol Pharmacol 82(11): 935-939, 2004. PMID: 15644932.
- Balbo S, Johnson CS, Kovi RC, James-Yi SA, O'Sullivan MG, Wang M, et al. Carcinogenicity and DNA adduct formation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in F-344 rats. Carcinogenesis 35(12): 2798-2806, 2014. PMID: 25269804.
- Banerjee J, Al-Wadei HA and Schuller HM. Chronic nicotine inhibits the therapeutic effects of gemcitabine on pancreatic cancer in vitro and in mouse xenografts. Eur J Cancer 49(5): 1152-1158, 2013. PMID: 23146955.
- Bastida G and Beltran B. Ulcerative colitis in smokers, non-smokers and ex-smokers. World J Gastroenterol 17(22): 2740-2747, 2011. PMID: 21734782.
- Blackford A, Parmigiani G, Kensler TW, Wolfgang C, Jones S, Zhang X, et al. Genetic mutations associated with cigarette smoking in pancreatic cancer. Cancer Res 69(8): 3681-3688, 2009. PMID: 19351817.
- Bourliere M, Barthet M, Berthezene P, Durbec JP and Sarles H. Is tobacco a risk factor for chronic pancreatitis and alcoholic cirrhosis? Gut 32(11): 1392-1395, 1991. PMID: 1752475.
- Cavestro GM, Leandro G, Di Leo M, Zuppardo RA, Morrow OB, Notaristefano C, et al. A single-centre prospective, cohort study of the natural history of acute pancreatitis. Dig Liver Dis 47(3): 205-210, 2015. PMID: 25475611.
- Chaudhuri A, Kolodecik TR and Gorelick FS. Effects of increased intracellular cAMP on carbachol-stimulated zymogen activation, secretion, and injury in the pancreatic acinar cell. Am J Physiol Gastrointest Liver Physiol 288(2): G235-243, 2005. PMID: 15458924.
- Chowdhury P. An exploratory study on the development of an animal model of acute pancreatitis following nicotine exposure. Tob Induc Dis 1(3): 213-217, 2003. PMID: 19570262.
- Chowdhury P, Bose C and Udupa KB. Nicotine-induced proliferation of isolated rat pancreatic acinar cells: effect on cell signalling and function. Cell Prolif 40(1): 125-141, 2007. PMID: 17227300.
- Chowdhury P, Doi R, Chang LW and Rayford PL. Tissue distribution of [3H]-nicotine in rats. Biomed Environ Sci 6(1): 59-64, 1993. PMID: 8476533.
- Chowdhury P, Doi R, Tangoku A and Rayford PL. Structural and functional changes of rat exocrine pancreas exposed to nicotine. Int J Pancreatol 18(3): 257-264, 1995. PMID: 8708398.
- Chowdhury P, Hosotani R, Chang L and Rayford PL. Metabolic and pathologic effects of nicotine on gastrointestinal tract and pancreas of rats. Pancreas 5(2): 222-229, 1990. PMID: 1690423.
- Chowdhury P, Hosotani R and Rayford PL. Inhibition of CCK or carbachol-stimulated amylase release by nicotine. Life Sci 45(22): 2163-2168, 1989. PMID: 2481202.
- Chowdhury P, Hosotani R and Rayford PL. Weight loss and altered circulating GI peptide levels of rats exposed chronically to nicotine. Pharmacol Biochem Behav 33(3): 591-594, 1989. PMID: 2587602.
- Chowdhury P, MacLeod S, Udupa KB and Rayford PL. Pathophysiological effects of nicotine on the pancreas: an update. Exp Biol Med 227(7): 445-454, 2002. PMID: 12094008.
- Chowdhury P and Rayford PL. Smoking and pancreatic disorders. Eur J Gastroenterol Hepatol 12(8): 869-877, 2000. PMID: 10958214.
- Chowdhury P, Rayford PL and Chang LW. Induction of pancreatic acinar pathology via inhalation of nicotine. Proc Soc Exp Biol Med 201(2): 159-164, 1992. PMID: 1409731.
- Chowdhury P, Rayford PL and Chang LW. Pathophysiological effects of nicotine on the pancreas. Proc Soc Exp Biol Med 218(3): 168-173, 1998. PMID: 9648934.
- Chowdhury P and Udupa KB. Effect of nicotine on exocytotic pancreatic secretory response: role of calcium signaling. Tob Induc Dis 11(1): 1, 2013. PMID: 23327436.
- Chowdhury P and Udupa KB. Nicotine as a mitogenic stimulus for pancreatic acinar cell proliferation. World J Gastroenterol 12(46): 7428-7432, 2006. PMID: 17167829.
- Cote GA, Yadav D, Slivka A, Hawes RH, Anderson MA, Burton FR, et al. Alcohol and smoking as risk factors in an epidemiology study of patients with chronic pancreatitis. Clin Gastroenterol Hepatol 9(3): 266-273, 2011. PMID: 21029787.
- Dasgupta P, Rizwani W, Pillai S, Kinkade R, Kovacs M, Rastogi S, et al. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int J Cancer 124(1): 36-45, 2009. PMID: 18844224.
- DiMagno MJ, Spaete JP, Ballard DD, Wamsteker EJ and Saini SD. Risk models for post-endoscopic retrograde cholangiopancreatography pancreatitis (PEP): smoking and chronic liver disease are predictors of protection against PEP. Pancreas 42(6): 996-1003, 2013. PMID: 23532001.
- Ding YS, Zhang L, Jain RB, Jain N, Wang RY, Ashley DL, et al. Levels of tobacco-specific nitrosamines and polycyclic aromatic hydrocarbons in mainstream smoke from different tobacco varieties. Cancer Epidemiol Biomarkers Prev 17(12): 3366-3371, 2008. PMID: 19064552.
- Duell EJ, Casella DP, Burk RD, Kelsey KT and Holly EA. Inflammation, genetic polymorphisms in proinflammatory genes TNF-A, RANTES, and CCR5, and risk of pancreatic adenocarcinoma. Cancer Epidemiol Biomarkers Prev 15(4): 726-731, 2006. PMID: 16614115.
- Edderkaoui M, Grippo PJ, Ouhaddi Y, Benhaddou H, Xu S, Pinkerton K, et al. Mouse models of pancreatic cancer induced by chronic pancreatitis and smoking. Journal of Clinical Oncology 32(3). (Abstract) 2014.
- Edderkaoui M and Thrower E. Smoking and Pancreatic Disease. J Cancer Ther 4(10A): 34-40, 2013. PMID: 24660091.
- Esposito I, Menicagli M, Funel N, Bergmann F, Boggi U, Mosca F, et al. Inflammatory cells contribute to the generation of an angiogenic phenotype in pancreatic ductal adenocarcinoma. J Clin Pathol 57(6): 630-636, 2004. PMID: 15166270.
- Foster JR, Idle JR, Hardwick JP, Bars R, Scott P and Braganza JM. Induction of drug-metabolizing enzymes in human pancreatic cancer and chronic pancreatitis. J Pathol 169(4): 457-463, 1993. PMID: 8501544.
- Franco L, Doria D, Bertazzoni E, Benini A and Bassi C. Increased expression of inducible nitric oxide synthase and cyclooxygenase-2 in pancreatic cancer. Prostaglandins Other Lipid Mediat 73(1-2): 51-58, 2004. PMID: 15165031.
- Friess H, Shrikhande S, Riesle E, Kashiwagi M, Baczako K, Zimmermann A, et al. Phospholipase A2 isoforms in acute pancreatitis. Ann Surg 233(2): 204-212, 2001. PMID: 11176126.
- Gnoni A, Licchetta A, Scarpa A, Azzariti A, Brunetti AE, Simone G, et al. Carcinogenesis of pancreatic adenocarcinoma: precursor lesions. Int J Mol Sci 14(10): 19731-19762, 2013. PMID: 24084722.
- Goecke H, Forssmann U, Uguccioni M, Friess H, Conejo-Garcia JR, Zimmermann A, et al. Macrophages infiltrating the tissue in chronic pancreatitis express the chemokine receptor CCR5. Surgery 128(5): 806-814, 2000. PMID: 11056444.
- Greer JB and Whitcomb DC. Inflammation and pancreatic cancer: an evidence-based review. Curr Opin Pharmacol 9(4): 411-418, 2009. PMID: 19589727.
- Haritha J and Wilcox CM. Evaluation of Patients' Knowledge Regarding Smoking and Chronic Pancreatitis: A Pilot Study. J Gastroenterol Pancreatol Liver Disord 1(2): 1-4, 2015. PMID: 25685852.
- Hartwig W, Werner J, Ryschich E, Mayer H, Schmidt J, Gebhard MM, et al. Cigarette smoke enhances ethanol-induced pancreatic injury. Pancreas 21(3): 272-278, 2000. PMID: 11039472.
- Hecht SS. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem Res Toxicol 11(6): 559-603, 1998. PMID: 9625726.
- Hermann PC, Sancho P, Canamero M, Martinelli P, Madriles F, Michl P, et al. Nicotine promotes initiation and progression of KRAS-induced pancreatic cancer via Gata6-dependent dedifferentiation of acinar cells in mice. Gastroenterology 147(5): 1119-1133 e1114, 2014. PMID: 25127677.
- Hirota M, Shimosegawa T, Masamune A, Kikuta K, Kume K, Hamada S, et al. The seventh nationwide epidemiological survey for chronic pancreatitis in Japan: clinical significance of smoking habit in Japanese patients. Pancreatology 14(6): 490-496, 2014. PMID: 25224249.
- Iodice S, Gandini S, Maisonneuve P and Lowenfels AB. Tobacco and the risk of pancreatic cancer: a review and meta-analysis. Langenbecks Arch Surg 393(4): 535-545, 2008. PMID: 18193270.
- Jaster R and Emmrich J. Crucial role of fibrogenesis in pancreatic diseases. Best Pract Res Clin Gastroenterol 22(1): 17-29, 2008. PMID: 18206810.
- Je DW, O YM, Ji YG, Cho Y and Lee DH. The inhibition of SRC family kinase suppresses pancreatic cancer cell proliferation, migration, and invasion. Pancreas 43(5): 768-776, 2014. PMID: 24763074.
- Jianyu H, Guang L and Baosen p. Evidence for cigarette smoke-induced oxidative stress in the rat pancreas. Inhal Toxicol 21(12): 1007-1012, 2009. PMID: 19635036.
- Kadiyala V, Lee LS, Banks PA, Suleiman S, Paulo JA, Wang W, et al. Cigarette smoking impairs pancreatic duct cell bicarbonate secretion. JOP 14(1): 31-38, 2013. PMID: 23306332.
- Krasinskas AM, Chiosea SI, Pal T and Dacic S. KRAS mutational analysis and immunohistochemical studies can help distinguish pancreatic metastases from primary lung adenocarcinomas. Mod Pathol 27(2): 262-270, 2014. PMID: 23887294.
- Kumar S, Torres MP, Kaur S, Rachagani S, Joshi S, Johansson SL, et al. Smoking accelerates pancreatic cancer progression by promoting differentiation of MDSCs and inducing HB-EGF expression in macrophages. Oncogene 34(16): 2052-2060, 2015. PMID: 24909166.
- Larsen S, Hilsted J, Tronier B and Worning H. Pancreatic hormone secretion in chronic pancreatitis without residual beta-cell function. Acta Endocrinol (Copenh) 118(3): 357-364, 1988. PMID: 2899369.
- LaRusch J, Lozano-Leon A, Stello K, Moore A, Muddana V, O'Connell M, et al. The Common Chymotrypsinogen C (CTRC) Variant G60G (C.180T) Increases Risk of Chronic Pancreatitis But Not Recurrent Acute Pancreatitis in a North American Population. Clin Transl Gastroenterol 6: e68, 2015. PMID: 25569187.
- Law R, Parsi M, Lopez R, Zuccaro G and Stevens T. Cigarette smoking is independently associated with chronic pancreatitis. Pancreatology 10(1): 54-59, 2010. PMID: 20332662.
- Lazar M, Sullivan J, Chipitsyna G, Aziz T, Salem AF, Gong Q, et al. Induction of monocyte chemoattractant protein-1 by nicotine in pancreatic ductal adenocarcinoma cells: role of osteopontin. Surgery 148(2): 298-309, 2010. PMID: 20579680.
- Lin HH, Chang HY, Chiang YT, Wu MS, Lin JT and Liao WC. Smoking, drinking, and pancreatitis: a population-based cohort study in Taiwan. Pancreas 43(7): 1117-1122, 2014. PMID: 25083998.
- Lin Y, Tamakoshi A, Hayakawa T, Ogawa M and Ohno Y. Cigarette smoking as a risk factor for chronic pancreatitis: a case-control study in Japan. Research Committee on Intractable Pancreatic Diseases. Pancreas 21(2): 109-114, 2000. PMID: 10975702.
- Lindkvist B, Wierup N, Sundler F and Borgstrom A. Long-term nicotine exposure causes increased concentrations of trypsinogens and amylase in pancreatic extracts in the rat. Pancreas 37(3): 288-294, 2008. PMID: 18815551.
- Lowenfels AB and Maisonneuve P. Environmental factors and risk of pancreatic cancer. Pancreatology 3(1): 1-7, 2003. PMID: 12683400.
- Macha MA, Rachagani S, Gupta S, Pai P, Ponnusamy MP, Batra SK, et al. Guggulsterone decreases proliferation and metastatic behavior of pancreatic cancer cells by modulating JAK/STAT and Src/FAK signaling. Cancer Lett 341(2): 166-177, 2013. PMID: 23920124.
- Mack TM, Yu MC, Hanisch R and Henderson BE. Pancreas cancer and smoking, beverage consumption, and past medical history. J Natl Cancer Inst 76(1): 49-60, 1986. PMID: 3455742.
- Maire F, Rebours V, Vullierme MP, Couvelard A, Levy P, Hentic O, et al. Does tobacco influence the natural history of autoimmune pancreatitis? Pancreatology 14(4): 284-288, 2014. PMID: 25062878.
- Maisonneuve P and Lowenfels AB. Epidemiology of pancreatic cancer: an update. Dig Dis 28(4-5): 645-656, 2010. PMID: 21088417.
- Maisonneuve P, Lowenfels AB, Mullhaupt B, Cavallini G, Lankisch PG, Andersen JR, et al. Cigarette smoking accelerates progression of alcoholic chronic pancreatitis. Gut 54(4): 510-514, 2005. PMID: 15753536.
- Martinelli P, Madriles F, Canamero M, Pau EC, Pozo ND, Guerra C, et al. The acinar regulator Gata6 suppresses KrasG12V-driven pancreatic tumorigenesis in mice. Gut, 2015. PMID: 25596178.
- McGrath J, McDonald JW and Macdonald JK. Transdermal nicotine for induction of remission in ulcerative colitis. Cochrane Database Syst Rev(4): CD004722, 2004. PMID: 15495126.
- Milnerowicz H, Sliwinska-Mosson M, Rabczynski J, Nowak M and Milnerowicz S. Dysfunction of the pancreas in healthy smoking persons and patients with chronic pancreatitis. Pancreas 34(1): 46-54, 2007. PMID: 17198182.
- Momi N, Ponnusamy MP, Kaur S, Rachagani S, Kunigal SS, Chellappan S, et al. Nicotine/cigarette smoke promotes metastasis of pancreatic cancer through alpha7nAChR-mediated MUC4 upregulation. Oncogene 32(11): 1384-1395, 2013. PMID: 22614008.
- Mule JJ, Custer M, Averbook B, Yang JC, Weber JS, Goeddel DV, et al. RANTES secretion by gene-modified tumor cells results in loss of tumorigenicity in vivo: role of immune cell subpopulations. Hum Gene Ther 7(13): 1545-1553, 1996. PMID: 8864755.
- Mwenifumbo JC and Tyndale RF. Molecular genetics of nicotine metabolism. Handb Exp Pharmacol(192): 235-259, 2009. PMID: 19184652.
- Park CH, Lee IS, Grippo P, Pandol SJ, Gukovskaya AS and Edderkaoui M. Akt kinase mediates the prosurvival effect of smoking compounds in pancreatic ductal cells. Pancreas 42(4): 655-662, 2013. PMID: 23271397.
- Paulo JA. Nicotine alters the proteome of two human pancreatic duct cell lines. JOP 15(5): 465-474, 2014. PMID: 25262714.
- Paulo JA, Urrutia R, Kadiyala V, Banks P, Conwell DL and Steen H. Cross-species analysis of nicotine-induced proteomic alterations in pancreatic cells. Proteomics 13(9): 1499-1512, 2013. PMID: 23456891.
- Philip B, Roland CL, Daniluk J, Liu Y, Chatterjee D, Gomez SB, et al. A high-fat diet activates oncogenic Kras and COX2 to induce development of pancreatic ductal adenocarcinoma in mice. Gastroenterology 145(6): 1449-1458, 2013. PMID: 23958541.
- Porta M, Crous-Bou M, Wark PA, Vineis P, Real FX, Malats N, et al. Cigarette smoking and K-ras mutations in pancreas, lung and colorectal adenocarcinomas: etiopathogenic similarities, differences and paradoxes. Mutat Res 682(2-3): 83-93, 2009. PMID: 19651236.
- Prizment AE, Jensen EH, Hopper AM, Virnig BA and Anderson KE. Risk factors for pancreatitis in older women: the Iowa Women's Health Study. Ann Epidemiol, 2015. PMID: 25656921.
- Prokopczyk B, Hoffmann D, Bologna M, Cunningham AJ, Trushin N, Akerkar S, et al. Identification of tobacco-derived compounds in human pancreatic juice. Chem Res Toxicol 15(5): 677-685, 2002. PMID: 12018989.
- Raimondi S, Lowenfels AB, Morselli-Labate AM, Maisonneuve P and Pezzilli R. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol 24(3): 349-358, 2010. PMID: 20510834.
- Raimondi S, Maisonneuve P, Lohr JM and Lowenfels AB. Early onset pancreatic cancer: evidence of a major role for smoking and genetic factors. Cancer Epidemiol Biomarkers Prev 16(9): 1894-1897, 2007. PMID: 17855711.
- Rebours V, Vullierme MP, Hentic O, Maire F, Hammel P, Ruszniewski P, et al. Smoking and the Course of Recurrent Acute and Chronic Alcoholic Pancreatitis A Dose-Dependent Relationship. Pancreas 41(8): 1219-1224, 2012. PMID: 23086245.
- Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and dissemination precede pancreatic tumor formation. Cell 148(1-2): 349-361, 2012. PMID: 22265420.
- Rioux N and Castonguay A. 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone modulation of cytokine release in U937 human macrophages. Cancer Immunol Immunother 49(12): 663-670, 2001. PMID: 11258792.
- Rivenson A, Hoffmann D, Prokopczyk B, Amin S and Hecht SS. Induction of lung and exocrine pancreas tumors in F344 rats by tobacco-specific and Areca-derived N-nitrosamines. Cancer Res 48(23): 6912-6917, 1988. PMID: 3180100.
- Rosenfeld G and Bressler B. The truth about cigarette smoking and the risk of inflammatory bowel disease. Am J Gastroenterol 107(9): 1407-1408, 2012. PMID: 22951878.
- Sadr-Azodi O, Andren-Sandberg A, Orsini N and Wolk A. Cigarette smoking, smoking cessation and acute pancreatitis: a prospective population-based study. Gut 61(2): 262-267, 2012. PMID: 21836026.
- Sasco AJ, Secretan MB and Straif K. Tobacco smoking and cancer: a brief review of recent epidemiological evidence. Lung Cancer 45 Suppl 2: S3-9, 2004. PMID: 15552776.
- Schneider L, Jabrailova B, Soliman H, Hofer S, Strobel O, Hackert T, et al. Pharmacological cholinergic stimulation as a therapeutic tool in experimental necrotizing pancreatitis. Pancreas 43(1): 41-46, 2014. PMID: 24212240.
- Schuller HM. Nitrosamines as nicotinic receptor ligands. Life Sci 80(24-25): 2274-2280, 2007. PMID: 17459420.
- Schuller HM, Tithof PK, Williams M and Plummer H, 3rd. The tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone is a beta-adrenergic agonist and stimulates DNA synthesis in lung adenocarcinoma via beta-adrenergic receptor-mediated release of arachidonic acid. Cancer Res 59(18): 4510-4515, 1999. PMID: 10493497.
- Shimizu K. Pancreatic stellate cells: molecular mechanism of pancreatic fibrosis. J Gastroenterol Hepatol 23 Suppl 1: S119-121, 2008. PMID: 18336654.
- Sliwinska-Mosson M, Milnerowicz H, Jablonowska M, Milnerowicz S, Nabzdyk S and Rabczynski J. The effect of smoking on expression of IL-6 and antioxidants in pancreatic fluids and tissues in patients with chronic pancreatitis. Pancreatology 12(4): 295-304, 2012. PMID: 22898629.
- Sliwinska-Mosson M, Milnerowicz H, Milnerowicz S, Nowak M and Rabczynski J. Immunohistochemical localization of somatostatin and pancreatic polypeptide in smokers with chronic pancreatitis. Acta Histochem 114(5): 495-502, 2012. PMID: 22113176.
- Sliwinska-Mosson M, Milnerowicz S, Nabzdyk S, Kokot I, Nowak M and Milnerowicz H. The Effect of Smoking on Endothelin-1 in Patients With Chronic Pancreatitis. Appl Immunohistochem Mol Morphol, 2014. PMID: 25203431.
- Sliwinska-Mosson M, Sciskalska M, Karczewska-Gorska P and Milnerowicz H. The effect of endothelin-1 on pancreatic diseases in patients who smoke. Adv Clin Exp Med 22(5): 745-752, 2013. PMID: 24285461.
- Sliwinska-Mosson M, Topola M, Milnerowicz S and Milnerowicz H. [Assessment of concentrations of creatinine, uric acid and urea in non-smoking and smoking patients with chronic pancreatitis]. Przegl Lek 70(10): 809-812, 2013. PMID: 24501801.
- Soeda J, Morgan M, McKee C, Mouralidarane A, Lin C, Roskams T, et al. Nicotine induces fibrogenic changes in human liver via nicotinic acetylcholine receptors expressed on hepatic stellate cells. Biochem Biophys Res Commun 417(1): 17-22, 2012. PMID: 22108052.
- Song Z, Bhagat G, Quante M, Baik GH, Marrache F, Tu SP, et al. Potential carcinogenic effects of cigarette smoke and Swedish moist snuff on pancreas: a study using a transgenic mouse model of chronic pancreatitis. Lab Invest 90(3): 426-435, 2010. PMID: 20065943.
- Srinivasan P, Subramanian VS and Said HM. Effect of the cigarette smoke component, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), on physiological and molecular parameters of thiamin uptake by pancreatic acinar cells. PLoS One 8(11): e78853, 2013. PMID: 24244374.
- Stocks P. Cancer mortality in relation to national consumption of cigarettes, solid fuel, tea and coffee. Br J Cancer 24(2): 215-225, 1970. PMID: 5451565.
- Talamini G, Bassi C, Falconi M, Sartori N, Salvia R, Rigo L, et al. Alcohol and smoking as risk factors in chronic pancreatitis and pancreatic cancer. Dig Dis Sci 44(7): 1303-1311, 1999. PMID: 10489910.
- Tolstrup JS, Kristiansen L, Becker U and Gronbaek M. Smoking and risk of acute and chronic pancreatitis among women and men: a population-based cohort study. Arch Intern Med 169(6): 603-609, 2009. PMID: 19307524.
- Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest 117(2): 289-296, 2007. PMID: 17273548.
- Trevino JG, Pillai S, Kunigal S, Singh S, Fulp WJ, Centeno BA, et al. Nicotine induces inhibitor of differentiation-1 in a Src-dependent pathway promoting metastasis and chemoresistance in pancreatic adenocarcinoma. Neoplasia 14(12): 1102-1114, 2012. PMID: 23308043.
- Trushin N, Leder G, El-Bayoumy K, Hoffmann D, Beger HG, Henne-Bruns D, et al. The tobacco carcinogen NNK is stereoselectively reduced by human pancreatic microsomes and cytosols. Langenbecks Arch Surg 393(4): 571-579, 2008. PMID: 18259773.
- van Geenen EJ, Smits MM, Schreuder TC, van der Peet DL, Bloemena E and Mulder CJ. Smoking is related to pancreatic fibrosis in humans. Am J Gastroenterol 106(6): 1161-1166; quiz 1167, 2011. PMID: 21577244.
- van Westerloo DJ, Giebelen IA, Florquin S, Bruno MJ, Larosa GJ, Ulloa L, et al. The vagus nerve and nicotinic receptors modulate experimental pancreatitis severity in mice. Gastroenterology 130(6): 1822-1830, 2006. PMID: 16697744.
- Vickers SM, MacMillan-Crow LA, Green M, Ellis C and Thompson JA. Association of increased immunostaining for inducible nitric oxide synthase and nitrotyrosine with fibroblast growth factor transformation in pancreatic cancer. Arch Surg 134(3): 245-251, 1999. PMID: 10088562.
- Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421(6921): 384-388, 2003. PMID: 12508119.
- Whittemore AS, Paffenbarger RS, Jr., Anderson K and Halpern J. Early precursors of pancreatic cancer in college men. J Chronic Dis 36(3): 251-256, 1983. PMID: 6826689.
- Wittel UA, Pandey KK, Andrianifahanana M, Johansson SL, Cullen DM, Akhter MP, et al. Chronic pancreatic inflammation induced by environmental tobacco smoke inhalation in rats. Am J Gastroenterol 101(1): 148-159, 2006. PMID: 16405548.
- Wittel UA, Singh AP, Henley BJ, Andrianifahanana M, Akhter MP, Cullen DM, et al. Cigarette smoke-induced differential expression of the genes involved in exocrine function of the rat pancreas. Pancreas 33(4): 364-370, 2006. PMID: 17079941.
- Wojcik J, Kuska J and Kokot F. [Effect of smoking on the secretion of immunoreactive insulin (IRI) induced by the administration of L-arginine or glucagon]. Endokrynol Pol 36(5): 263-269, 1985. PMID: 3913601.
- Wynder EL. An epidemiological evaluation of the causes of cancer of the pancreas. Cancer Res 35(8): 2228-2233, 1975. PMID: 1149034.
- Wynder EL, Mabuchi K, Maruchi N and Fortner JG. Epidemiology of cancer of the pancreas. J Natl Cancer Inst 50(3): 645-667, 1973. PMID: 4350660.
- Xu Z, Lee A, Pothula S, Patel M, Pirola R, Wilson J, et al. The influence of alcohol and cigarette smoke components on pancreatic stellate cell activation: Implications for the progression of chronic pancreatitis. Pancreatology 14(3): S18-S19. (Abstract) 2014.
- Yadav D, Hawes RH, Brand RE, Anderson MA, Money ME, Banks PA, et al. Alcohol consumption, cigarette smoking, and the risk of recurrent acute and chronic pancreatitis. Arch Intern Med 169(11): 1035-1045, 2009. PMID: 19506173.
- Yadav D, Slivka A, Sherman S, Hawes RH, Anderson MA, Burton FR, et al. Smoking is underrecognized as a risk factor for chronic pancreatitis. Pancreatology 10(6): 713-719, 2010. PMID: 21242712.
- Yang H, Wang L, Shi YH, Sui GT, Wu YF, Lu XQ, et al. Risk factors of acute pancreatitis in the elderly Chinese population: a population-based cross-sectional study. J Dig Dis 15(9): 501-507, 2014. PMID: 24957953.
- Yuhara H, Ogawa M, Kawaguchi Y, Igarashi M and Mine T. Smoking and risk for acute pancreatitis: a systematic review and meta-analysis. Pancreas 43(8): 1201-1207, 2014. PMID: 25333404.
- Zheng YS, Wu ZS, Zhang LY, Ke L, Li WQ, Li N, et al. Nicotine Ameliorates Experimental Severe Acute Pancreatitis via Enhancing Immunoregulation of CD4+ CD25+Regulatory T Cells. Pancreas 44(3): 500-506, 2015. PMID: 25742430.
- Zhou J and Sahin-Toth M. Chymotrypsin C mutations in chronic pancreatitis. J Gastroenterol Hepatol 26(8): 1238-1246, 2011. PMID: 21631589.