
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2016.25
| Attachment | Size |
|---|---|
| 501.58 KB |
Abstract:
Pancreatitis is an inflammatory disease that starts in pancreatic acinar cells and results in significant morbidity and mortality. Currently there is no therapy for pancreatitis because of lack of complete understanding of the disease mechanism. Premature trypsinogen activation is considered to be the key event in the disease development. However recent research in the field has pointed out that besides trypsinogen activation there are many other important factors that play important role in the disease. Our recent study in Trypsinogen-7 knock out (T7-/-) mice has supported this and for the first time shown that intra-acinar trypsinogen activation contributes only partially to acinar injury and local and systemic inflammation progress independently of trypsin activation during pancreatitis. NF-κB activation that happens parallel and independent to trypsinogen activation can still drive the development of the acute or chronic pancreatitis even in absence of trypsin.
1. Introduction
Pancreatitis is an inflammatory disease of the pancreas that starts in pancreatic acinar cells and results in significant morbidity and mortality (19). More than a century ago, pathologist Dr. Hans Chiari (6) proposed that the acute pancreatitis is a disease rather than an infection, in which pancreas destroy itself through autodigestion. Since then the mechanism, site and importance of premature activation of digestive enzymes, especially trypsin, has become a major area of investigation in pathobiology of pancreatitis. Premature activation of trypsin has been observed during both in vitro hyperstimulation and in animal models of acute pancreatitis (16, 17, 22-24). In this review, we have discussed our current understanding of the role of trypsin in the pathophysiology of pancreatitis.
2. Physiology of Trypsinogen in Health
Trypsin is synthesized as trypsinogen, an inactive precursor, in the rough endoplasmic reticulum and transported to the Golgi apparatus for sorting. Trypsinogen is always co- synthesized and packed with a pancreatic secretory trypsin inhibitor (PSTI) that inhibits its premature activation. Once, it reaches the Golgi system, the trypsinogen and other digestive enzymes condense into core particles and are packed in zymogen granules. The condensed enzymes are stable and minimal activation happens within the zymogen granules. Once acini receive secretory stimulus, these zymogen granules are released in to the lumen of pancreatic duct, which carries the digestive enzymes into the duodenum. Once in duodenum, enteropeptidase activates trypsinogen by removing 7-10 amino acid from N-terminal region known as trypsinogen activation peptide (TAP). Removal of TAP induces conformational change resulting in active trypsin. TAP is immunologically distinct from the same sequence within trypsinogen, thereby allowing detection of trypsinogen activation in situ (1, 10).
3. Intra-acinar Location of Trypsin Activation During Acute Pancreatitis
While it is clear that there is intra-acinar activation of trypsin, the exact intracellular location where trypsin is activated is a hotly debated area in pancreatitis. Subcellular fractionation and analysis of pancreatic homogenate shortly after the induction of pancreatitis have provided much insight about the location of trypsinogen activation. In vitro studies on acinar cells show that within 30 min of hyper stimulation most of the active trypsin localizes to a heavy, zymogen-rich pellet and after 60 min of hyperstimulation, the trypsin activity shifts to the supernatant. This shift was paralleled by appearance of immunoreactive TAP and cathepsin B, a lysosomal enzyme capable of activating trypsinogen, in the soluble fraction (14). This experimental evidence led to development of the “co-localization hypothesis” which purports that during acute pancreatitis lysosomal enzymes and zymogens fuse to form structures named co-localization organelles. Further, it has been proposed that in these co-localization organelles lysosomal enzyme cathepsin B activates trypsinogen to trypsin. Studies have shown that premature trypsinogen activation occurs in membrane-bound compartments resembling autophagic vesicles formed by co-localization of zymogen and lysosomes (22). In these co-localized vacuoles, the lysosomal protease cathepsin B activates trypsinogen. It is proposed that the active trypsin further activates other digestive enzymes with in acinar cells, presumably in the same manner, as it normally would do in the duodenum. The presence of these co-localization vacuoles has been observed in all models of experimental pancreatitis as wells as in pathological specimens of human pancreatitis. The studies, which have demonstrated that cathepsin B can activate trypsinogen in vitro, further support this theory (25, 29) and it seems that in the absence of cathepsin B, less than 1 % of the trypsinogen peptide is hydrolyzed but after incubation with cathepsin B for 30 min at pH 5, 96% of trypsinogen peptide was hydrolyzed (29). Interestingly, cathepsin B mediated trypsinogen activation seems not to be a crucial pathogenic step in hereditary pancreatitis patients with the trypsinogen mutations D22G and K23R (29).
4. Role of Trypsin During Acute Pancreatitis
For decades, intra-acinar trypsin activation has been considered the key event in acute pancreatitis. This trypsin centric hypothesis has been supported by various observations. Inhibition of trypsin, by somewhat nonspecific protease inhibitors, provides protection against injury during acute pancreatitis (31, 32). Furthermore, inhibition of trypsinogen activation by inhibiting the activity of cathepsin B or by deleting the cathepsin B gene also decreases pancreatic injury during acute pancreatitis, again suggesting that trypsinogen activation is important for pancreatic damage (12, 31). Halangk et al (12) showed that cathepsin B knock out (CB-KO) mice have less necrosis when compared to WT type mice. However, the degree of leukocyte infiltration in the pancreas or lungs during pancreatitis was not affected by the absence of cathepsin B indicating cathepsin B, and thus trypsin, independent evolution of local and systemic inflammation.
The strongest support for the trypsin centric theory comes from identification of mutations in the cationic trypsinogen gene PRSS1 in hereditary pancreatitis, an uncommon form of pancreatitis with autosomal dominant inheritance (34)(M, 2006 #17;Teich N, 2002 #11). The biochemical studies of the pancreatitis-associated p.R122H mutations of human cationic trypsinogen in vitro show that this trypsinogen variant has an increased propensity for auto-activation and is resistant to degradation by chymotrypsin C (30). However, this mutation has pleiotropic effects rather than being exclusively an activating mutation, and there is no direct evidence for increased intracellular trypsin activity in patients with hereditary pancreatitis due to this mutation (3). Furthermore, patients with this mutation do not suffer continuous pancreatitis but rather experience episodic attacks of the disease. Also, in mouse model of hereditary pancreatitis generated by transgenic expression of R122H trypsinogen, no increased trypsinogen activation was observed indicating involvement of other factors. R122H mutation has been shown to increase the frequency of trypsin auto-activation (3, 9, 15). Study of another PRSS1 mutant p.R116C deciphered an entirely novel mechanism of acinar cell injury, which is unrelated to trypsinogen activation. The mutation induces proenzyme misfolding leading to endoplasmic reticulum (ER) stress and unfolded protein response (UPR) activation (15). The trypsinogen activation hypothesis of hereditary pancreatitis also does not explain incomplete penetrance, the intermittent nature of the disease, and lack of progression to chronic pancreatitis in some individuals despite recurrent episodes. While, emerging epidemiologic and genetic data continue to link pancreatitis to trypsinogen activation, it is becoming increasingly clear that with the exception of hereditary pancreatitis and cystic fibrosis, a direct simplistic genetic mechanism for acute pancreatitis may not exist but instead, a complex interplay between genetic, environmental and developmental factors govern the susceptibility, progression and severity of pancreatitis.
Evaluating the role of trypsin activation, Gaiser et al (9) showed that expression of active trypsin in pancreas was sufficient for induction of acute pancreatitis. This study showed that moderate to low constitutive expression of rat anionic trypsinogen PRSS2 in acini, was sufficient to induce pancreatitis. Though the study diverged from the known pattern of transient but high-level trypsin activation, which is an important limitation of this model, the conclusions from this model again supported the role of trypsin activation in pathogenesis of pancreatitis (9). However, this overexpression model is somewhat artificial and lacks the stimuli and other intra-acinar processes observed during acute pancreatitis. Contrary to these findings, a study by Wartmann et al reported that cathepsin L knockout mice have much higher trypsinogen activation but significantly reduced pancreatic injury suggesting trypsinogen activation may even have a protective role during pancreatitis by degrading trypsinogen and other proteases (33).
To obtain further insight into the role of trypsin in acute pancreatitis, we have generated a novel knockout mouse lacking trypsinogen isoform-7 (mouse paralog of human cationic trypsinogen, PRSS1) (7). In this mouse strain, we do not observe intra-acinar pathologic trypsin activation during acute pancreatitis suggesting that the trypsinogen isoform-7 (T7) is responsible for the pathologic trypsin activation. Intriguingly, in these novel trypsinogen-7 knockout mice (T-/-) mice we observed that the acinar cell necrosis during caerulein (Figure 1A) as well as L-arginine induced acute pancreatitis is reduced to about half of that observed in mice with intact T7 (7). In vitro we observe that acini lacking T7 do not undergo necrosis (as measured by LDH release) when stimulated by supramaximal dose of caerulein. Furthermore, we observed that NF-κB activation as well as local or systemic inflammation is not altered by absence of trypsin (Figure 1 B and C) (7). These together suggest that trypsin is only partly responsible for acinar cell necrosis observed during acute pancreatitis and local and systemic inflammation is independent of trypsin. One could also extrapolate this to suggest that inflammatory cells and mediators are responsible for acinar cell injury observed in the absence of trypsin. The fact that markers of local and systemic injury during pancreatitis were not affected by the absence of trypsinogen activation underscores the importance of so far under-appreciated trypsin-independent events in acute pancreatitis (13). Therefore, it is likely that trypsin is only required for initiation of the injury and trypsin-independent inflammatory pathways (importantly NF-κB) determine disease progression and severity.
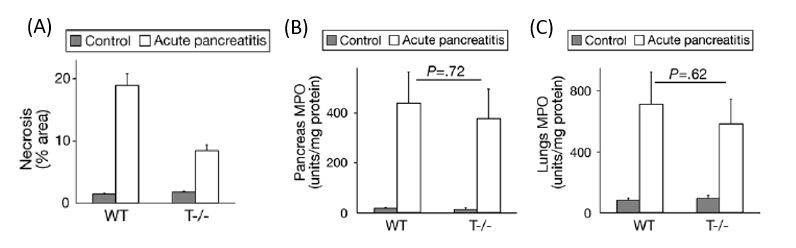
Figure 1: Trypsin contributes partially to acinar cell necrosis during Acute pancreatitis. Local and systemic inflammation during acute pancreatitis are independent of trypsin activation. Acute pancreatitis was induced by repeated injections caerulein (50 μg/kg i.p. every hour for 10 hours). A) Quantification of necrosis by morphometry. B) Quantification of Neutrophil infiltration (MPO) as a measure of inflammation in pancreas. C) lung MPO as a measure of inflammation in the lungs. Modified from Dawra et al (7)
5. Mechanism by Which Trypsin Leads to Acinar Cell Injury
We recently evaluated the mechanism by which trypsin induces cell death in acinar cells and observed that trypsin makes co-localized vesicles fragile which causes cathepsin B to escape from co-localized organelles into the cytosol, which in turn cause cell death during pancreatitis (28). Supramaximal stimulation by caerulein causes leakage of cathepsin B into cytoplasm (Figure 2A) and this release is dependent on trypsin as in its absence, either in T-/- mice or by pharmacologic inhibition of trypsin (Figure 2B), the release of cathepsin B into the cytosol during acute pancreatitis was prevented. These data suggest that active trypsin within the co-localized organelles plays a role in making the membranes fragile, through which the cathepsin B and other enzymes leak out into the cytosol. Furthermore, only a fraction of cathepsin B, amylase, active trypsin and arylsulfatase are released from the co-localized organelles into the cytosol. This suggests, as observed by us previously, that only a portion of lysosomes and zymogen granules come together and co-localize. The factors that determine which co-localized organelles become leaky and release their content into the cytosol are not known and are the area of future investigations.
Supramaximal caerulein stimulation leads to apoptosis that can be prevented by pretreatment with cathepsin B and trypsin inhibitors suggesting role of both cathepsin B and trypsin in acinar cell apoptosis (Figure 2C). However, when cathepsin B or trypsin was added to the permeabilized acini, to simulate presence of cathepsin B or trypsin in the cytosol, dose dependent activation of apoptosis was seen with the presence of cytosolic cathepsin B but not trypsin (Figure 2D) (28). This suggests the role of cytosolic cathepsin B but not trypsin in inducing acinar cell apoptosis. These observations have been conclusively supported by similar findings from experiments using T-/- and cathepsin B KO animals (28). From these studies, the most logical inference is that active trypsin within the co-localized organelles is involved in making the organelles “leaky,” causing leakage of cathepsin B into the cytosol, where the newly released cathepsin B activates apoptotic pathways. Inhibition of trypsin prevents the co-localized organelles from becoming fragile, thereby preventing the release of cathepsin B into the cytosol. Exogenous trypsin failed to activate caspase when incubated with streptolysin-O-permeabilized acinar cells suggesting trypsin does not directly cause acinar cell death.

Figure 2: A) Immuno-blot demonstrating that supramaximal caerulein stimulation leads to increase in cytosolic cathepsin B in acinar cells B) Cytosolic cathepsin B activity normalized to LDH is increased in pancreatic acinar cell treated with supramaximal dose of caerulein. However, similar increase is not observed in T-/- mice suggesting trypsin activity is required for release of cathepsin B into the cytosol C) Caspase-3 activity in acinar cell treated with supra maximal caerulein alone or caerulein with CA047 or caerulein with Benzamidine. D) Addition of exogenous cathepsin B but not trypsin to SLO permeabilized normal rat pancreatic acinar cells leads to caspase-3 activation. Caspase-3 activity was induced by cathepsin B in concentration dependent manner and no activity was seen with any given dose of trypsin. Values are expressed as percent of total and normalized to per mg protein. Modified from Talukdar et al (28)
There are two major pathways through which apoptosis occurs 1) extrinsic and 2) intrinsic pathway. The extrinsic pathway involves death receptors and occurs in response to external signals whereas the intrinsic pathway involves mitochondria and happens in response to internal signals. Lysosomal disruption has been implicated in initiating the intrinsic apoptotic pathway involving cleavage of pro-apoptotic Bcl-2 family member Bid. Upon apoptotic stimuli, Bcl-2 apoptosis-promoting protein Bax undergoes a conformational change and translocates to mitochondria, where it oligomerizes and forms pores that allow the release of cytochrome c into cytoplasm. It has also been shown that in early stages of experimental acute pancreatitis, there is a release of cytochrome c into the cytosol that in turn activates caspase-9, which subsequently leads to caspase-3 activation. Caspase-3 then executes the intracellular changes of apoptosis via different downstream mediators (8).
Our studies suggest that during acinar cell death, cathepsin B is releases into the cytosol and induces apoptosis predominantly via the intrinsic pathway by inducing Bid cleavage and Bax activation. Truncated Bid and activated Bax cause release of cytochrome c from mitochondria, which in turn leads to caspase-3 activation and acinar cell apoptosis. This cathepsin B induced apoptosis was fully inhibited in acini pretreated with CA074-me (Figure 2C). Moreover, the reduction of cytosolic cytochrome c after pre-treatment with cytochrome c antibody reduced cathepsin B triggered acinar cell apoptosis suggesting apoptosis via the intrinsic pathway (28).
Interestingly, the amount of cathepsin B in the cytosol determines whether acinar cell die via apoptosis or necrosis. Small amount of cathepsin B activates apoptosis whereas larger amounts of cathepsin B shift the cell death pathway towards necrosis. Necrosis is a form of cell death where there is swelling of organelles and plasma membrane rupture. Necrosis was once considered accidental cell death caused by overwhelming physical or chemical trauma. However, we now know that specific genes can induce necrosis in a regulated manner. The terms programmed necrosis, necroptosis, or regulated necrosis has been used to distinguish these types of cell death from accidental necrosis. Receptor-interacting protein 3 (RIP-3) is an important regulator of necroptosis. Once RIP-3 gets activated it forms a complex with receptor activating kinase 1(RIP-1). RIP-1/RIP-3 complex activates a cascade of events, which eventually lead to cell necrosis (18). The leakage of a large amount of cathepsin B by hyperstimulation of acinar cells leads to RIP-1/RIP-3 complex formation, which shifts the form of death from apoptosis to necrosis. In future, studies elucidation of the key molecules favoring the formation of RIP-1/RIP-3 complex and downstream events in the pathogenesis of acute pancreatitis may help decipher the mechanism of selection of apoptosis versus necrotic form of cell death during acute pancreatitis.
Besides premature trypsinogen activation, intra-acinar NF-κB activation has been previously shown to result in local pancreatic damage as well as systemic inflammation (4, 5). NF-κB related injury was persistent in acinar cell even in the absence of trypsin in T−/− mice suggesting its trypsin independent role in acinar cell injury. Although activation of other inflammatory cascades has been described in acute pancreatitis and may theoretically lead to trypsin independent and NF-κB independent injury, which need to explored in further studies, these pathways are generally known to be minor players compared to NF-κB pathway.
The soluble inflammatory cell mediators like TNF-α, which is a product of the NF-κB pathway, was shown to directly induce premature trypsinogen activation and necrosis in pancreatic acinar cells suggesting contribution of inflammatory signaling in disease initiation and progression (26). Thus NF-κB activation may also be the key early event responsible for progression of systemic injury. This fact was further supported by another important study, which demonstrated partial reduction in acinar necrosis during pancreatitis in mice lacking the p50 unit of NF-κB (p50−/−) (2). A study done by Gukovskaya et al (11) observed that neutrophils which are recruited during inflammation were able to activate trypsin in acinar cell and this was dependent on neutrophil NAPDH oxidase suggesting involvement of immune cells like neutrophil in further promoting acinar cell injury.
6. Role of Trypsin in Chronic Pancreatitis (CP):
Using our T-/- mice we have also explored the role of trypsin in CP (20). Using caerulein model of CP we found comparable levels of acinar cell damage and fibrosis (Figure 3) in T-/- and WT C57BL/6 mice suggesting trypsin independent activation of acinar necrosis and stellate cell activation in CP. We further looked at T cell infiltration and NF-κB activation (20). We found that it was comparable in both T -/- and WT. Taken together the data from these studies supports the fact that persistent inflammation, possibly driven by NF-κB dependent pathways, can lead to CP even in absence of trypsin. We further verified this in human CP samples and all sample tested showed high NF-κB activity (Figure 4).
Pancreatic acinar cells are protein-synthesizing machinery for secretory proteins and any stress might lead disturbance of cellular homeostasis and endoplasmic reticulum (ER) stress that leads to activation of evolutionary conserved unfolded protein response (UPR) pathways. A study from our group showed that during acinar cell injury there is ER stress and activation of UPR as seen by up regulation of UPR components like CHOP, ATF-4, GRP-78 and XBP-1 during caerulein induced CP. However, T-/- that lack intra-acinar trypsinogen activation showed comparable levels of ER stress and activation of UPR suggesting minimal role of trypsin in causing ER stress during CP (21).
In conclusion, different genetic mouse models of trypsin over expression or lacking trypsinogen activation have led to some exciting results that challenge the century-old trypsin-centered theory of pancreatitis. Our current understanding of the pathogenesis of acute pancreatitis is depicted in Figure 5. No doubt trypsin is important for disease initiation, however, its contribution to the disease mechanism is overestimated. In fact, the usage of trypsin protease inhibitors in clinical practice fails to provide any resolution for SAP patients (27). It is becoming increasingly clear that release of cathepsin B, NF-κB signaling and ER stress in acinar cells are all crucial to the pathogenesis of pancreatitis and could be important drug target for pancreatitis.
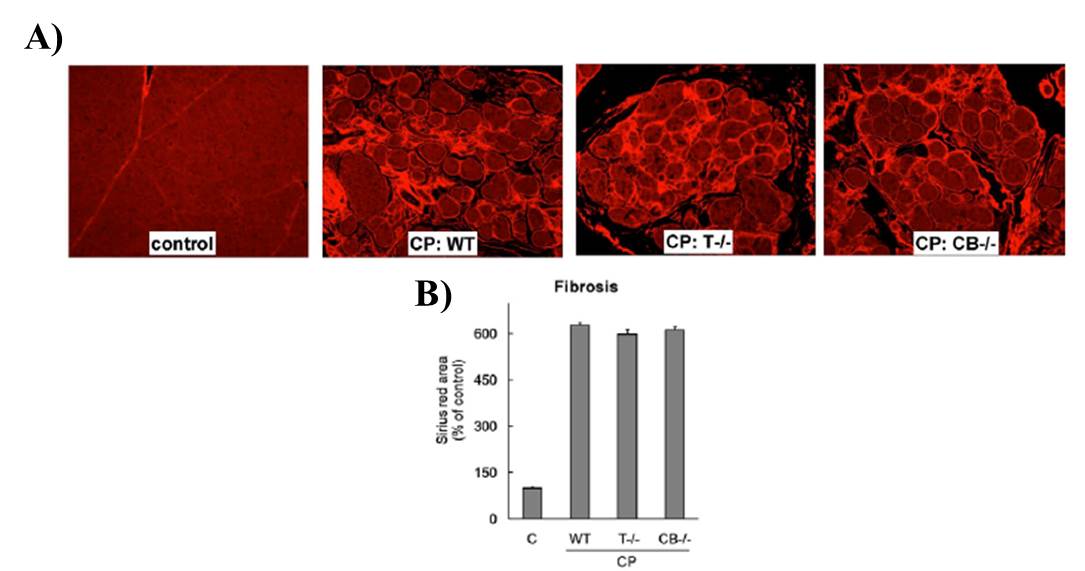
Figure 3: Chronic pancreatitis severity is not modulated by absence of trypsin. (A) Representative pictures of CP showing acinar fibrosis in WT, T-/-, and CB-/- CP groups. Fibrosis was detected by Sirius red–staining. (B) Quantification of Sirius red staining indicates comparable fibrosis in WT, T-/-, and CB-/- CP groups. Modified from Sah et al (20)
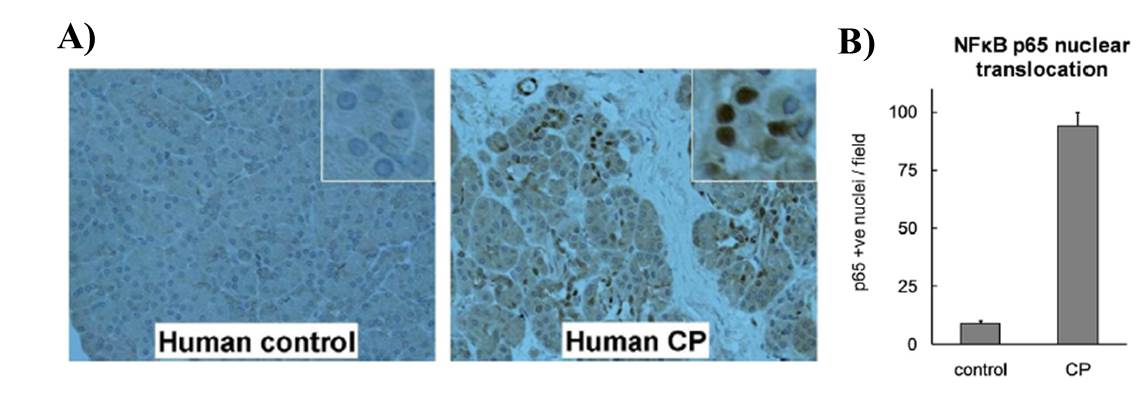
Figure 4: NF-kB activation in human chronic pancreatitis samples (A) Persistent NF-κB activation is observed in human chronic pancreatitis. NF-κB component protein p65 immunostaining shows its nuclear localization in acinar cells in human chronic pancreatitis sections (200X) (insets - zoomed-in views showing nucleus from acinar cells showing positive stain for p65). (B) The nuclear stain was quantified and represented here. Pancreas sections from 7 CP patients and 7 controls were analyzed. Modified from Sah et al (20)
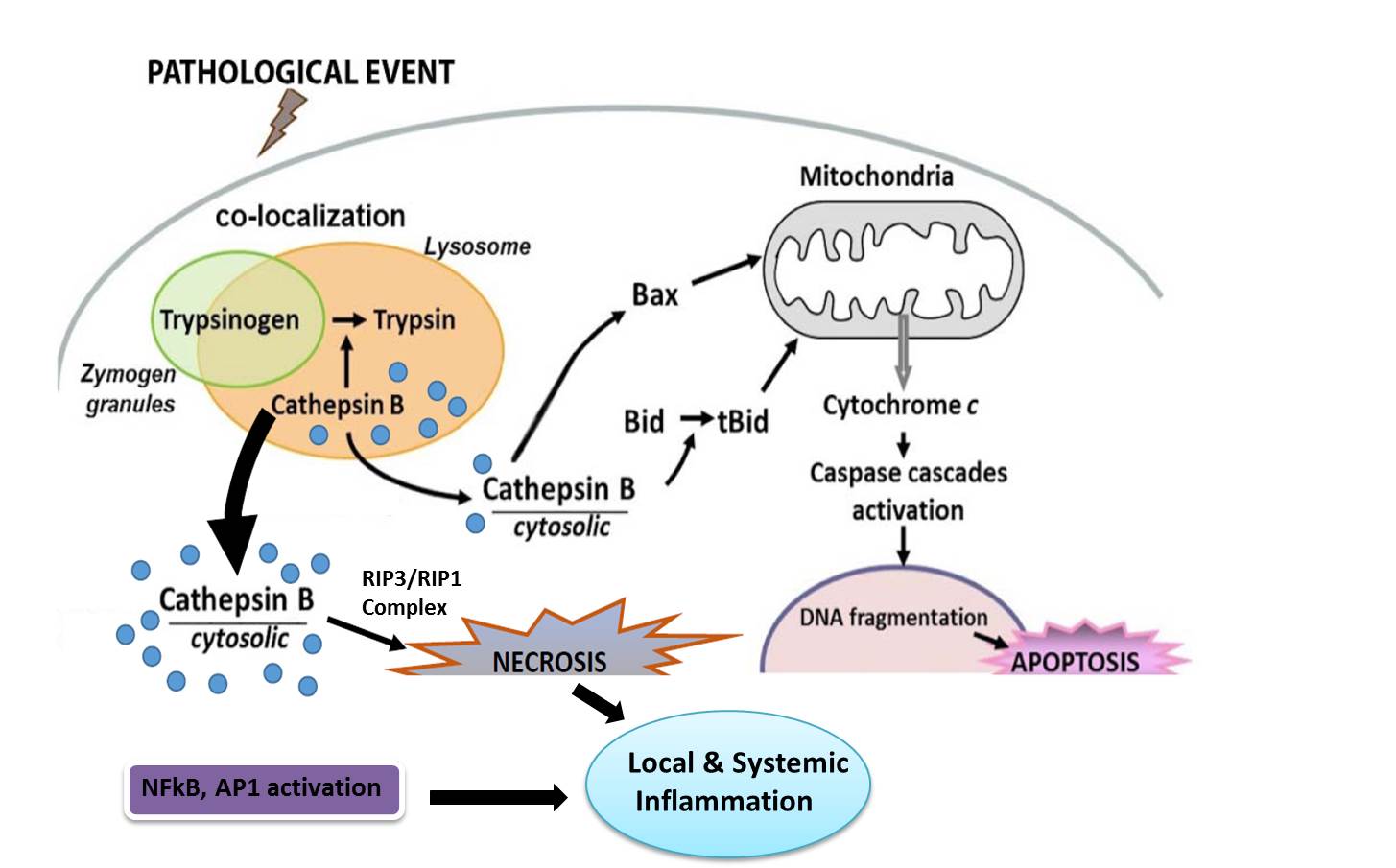
Figure 5: Schematic representation of major pathophysiologic events in pancreatitis.
7. References
- Abita JP,Delaage M and Lazdunski M. The mechanism of activation of trypsinogen. The role of the four N-terminal aspartyl residues. Eur J Biochem 8(3): 314-324, 1969. PMID: 5816755.
- Altavilla D,Famulari C,Passaniti M,Galeano M,Macri A,Seminara P, et al. Attenuated cerulein-induced pancreatitis in nuclear factor-kappaB-deficient mice. Lab Invest 83(12): 1723-1732, 2003. PMID: 14691290.
- Archer H,Jura N,Keller J,Jacobson M and Bar-Sagi D. A mouse model of hereditary pancreatitis generated by transgenic expression of R122H trypsinogen. Gastroenterology 131(6): 1844-1855, 2006. PMID: 17087933.
- Baumann B,Wagner M,Aleksic T,von Wichert G,Weber CK,Adler G, et al. Constitutive IKK2 activation in acinar cells is sufficient to induce pancreatitis in vivo. J Clin Invest 117(6): 1502-1513, 2007. PMID: 17525799.
- Chen X,Ji B,Han B,Ernst SA,Simeone D and Logsdon CD. NF-kappaB activation in pancreas induces pancreatic and systemic inflammatory response. Gastroenterology 122(2): 448-457, 2002. PMID: 11832459.
- Chiari H. U¨ber die Selbstverdauung des menschlichen Pankreas. Zeitschrift fu¨r Heilkunde 17: 69-96, 1896.
- Dawra R,Sah RP,Dudeja V,Rishi L,Talukdar R,Garg P, et al. Intra-acinar trypsinogen activation mediates early stages of pancreatic injury but not inflammation in mice with acute pancreatitis. Gastroenterology 141(6): 2210-2217 e2212, 2011. PMID: 21875495.
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 35(4): 495-516, 2007. PMID: 17562483.
- Gaiser S,Daniluk J,Liu Y,Tsou L,Chu J,Lee W, et al. Intracellular activation of trypsinogen in transgenic mice induces acute but not chronic pancreatitis. Gut 60(10): 1379-1388, 2011. PMID: 21471572.
- Go VLW,DiMagno EP,Gardner JD,Lebenthal E,Reber HA and Scheele GA. The Pancreas. Biology, Pathobiology, and Disease. NY, Raven Press. 2, 1993.
- Gukovskaya AS,Vaquero E,Zaninovic V,Gorelick FS,Lusis AJ,Brennan ML, et al. Neutrophils and NADPH oxidase mediate intrapancreatic trypsin activation in murine experimental acute pancreatitis. Gastroenterology 122(4): 974-984, 2002. PMID: 11910350.
- Halangk W,Lerch MM,Brandt-Nedelev B,Roth W,Ruthenbuerger M,Reinheckel T, et al. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J Clin Invest 106(6): 773-781, 2000. PMID: 10995788.
- Hietaranta AJ,Saluja AK,Bhagat L,Singh VP,Song AM and Steer ML. Relationship between NF-kappaB and trypsinogen activation in rat pancreas after supramaximal caerulein stimulation. Biochem Biophys Res Commun 280(1): 388-395, 2001. PMID: 11162528.
- Hofbauer B,Saluja AK,Lerch MM,Bhagat L,Bhatia M,Lee HS, et al. Intra-acinar cell activation of trypsinogen during caerulein-induced pancreatitis in rats. Am J Physiol 275(2 Pt 1): G352-362, 1998. PMID: 9688663.
- Kereszturi E,Szmola R,Kukor Z,Simon P,Weiss FU,Lerch MM, et al. Hereditary pancreatitis caused by mutation-induced misfolding of human cationic trypsinogen: a novel disease mechanism. Hum Mutat 30(4): 575-582, 2009. PMID: 19191323.
- Lerch MM and Gorelick FS. Early trypsinogen activation in acute pancreatitis. Med Clin North Am 84(3): 549-563, viii, 2000. PMID: 10872413.
- Mithofer K,Fernandez-del Castillo C,Rattner D and Warshaw AL. Subcellular kinetics of early trypsinogen activation in acute rodent pancreatitis. Am J Physiol 274(1 Pt 1): G71-79, 1998. PMID: 9458775.
- Moriwaki K and Chan FK. RIP3: a molecular switch for necrosis and inflammation. Genes Dev 27(15): 1640-1649, 2013. PMID: 23913919.
- Pandol SJ,Saluja AK,Imrie CW and Banks PA. Acute pancreatitis: bench to the bedside. Gastroenterology 132(3): 1127-1151, 2007. PMID: 17383433.
- Sah RP,Dudeja V,Dawra RK and Saluja AK. Cerulein-induced chronic pancreatitis does not require intra-acinar activation of trypsinogen in mice. Gastroenterology 144(5): 1076-1085 e1072, 2013. PMID: 23354015.
- Sah RP,Garg SK,Dixit AK,Dudeja V,Dawra RK and Saluja AK. Endoplasmic reticulum stress is chronically activated in chronic pancreatitis. J Biol Chem 289(40): 27551-27561, 2014. PMID: 25077966.
- Saluja A,Hashimoto S,Saluja M,Powers RE,Meldolesi J and Steer ML. Subcellular redistribution of lysosomal enzymes during caerulein-induced pancreatitis. Am J Physiol 253(4 Pt 1): G508-516, 1987. PMID: 2821825.
- Saluja A,Saluja M,Villa A,Leli U,Rutledge P,Meldolesi J, et al. Pancreatic duct obstruction in rabbits causes digestive zymogen and lysosomal enzyme colocalization. J Clin Invest 84(4): 1260-1266, 1989. PMID: 2477393.
- Saluja AK,Bhagat L,Lee HS,Bhatia M,Frossard JL and Steer ML. Secretagogue-induced digestive enzyme activation and cell injury in rat pancreatic acini. Am J Physiol 276(4 Pt 1): G835-842, 1999. PMID: 10198325.
- Saluja AK,Donovan EA,Yamanaka K,Yamaguchi Y,Hofbauer B and Steer ML. Cerulein-induced in vitro activation of trypsinogen in rat pancreatic acini is mediated by cathepsin B. Gastroenterology 113(1): 304-310, 1997. PMID: 9207291.
- Sendler M,Dummer A,Weiss FU,Kruger B,Wartmann T,Scharffetter-Kochanek K, et al. Tumour necrosis factor alpha secretion induces protease activation and acinar cell necrosis in acute experimental pancreatitis in mice. Gut 62(3): 430-439, 2013. PMID: 22490516.
- Singh VP and Chari ST. Protease inhibitors in acute pancreatitis: lessons from the bench and failed clinical trials. Gastroenterology 128(7): 2172-2174, 2005. PMID: 15940654.
- Talukdar R,Sareen A,Zhu H,Yuan Z,Dixit A,Cheema H, et al. Release of cathepsin-B in cytosol causes cell death in acute pancreatitis. Gastroenterology, 2016. PMID: 27519471.
- Teich N,Bodeker H and Keim V. Cathepsin B cleavage of the trypsinogen activation peptide. BMC Gastroenterol 2: 16, 2002. PMID: 12102727.
- Teich N,Rosendahl J,Toth M,Mossner J and Sahin-Toth M. Mutations of human cationic trypsinogen (PRSS1) and chronic pancreatitis. Hum Mutat 27(8): 721-730, 2006. PMID: 16791840.
- Van Acker GJ,Saluja AK,Bhagat L,Singh VP,Song AM and Steer ML. Cathepsin B inhibition prevents trypsinogen activation and reduces pancreatitis severity. Am J Physiol Gastrointest Liver Physiol 283(3): G794-800, 2002. PMID: 12181196.
- Van Acker GJ,Weiss E,Steer ML and Perides G. Cause-effect relationships between zymogen activation and other early events in secretagogue-induced acute pancreatitis. Am J Physiol Gastrointest Liver Physiol 292(6): G1738-1746, 2007. PMID: 17332471.
- Wartmann T,Mayerle J,Kahne T,Sahin-Toth M,Ruthenburger M,Matthias R, et al. Cathepsin L inactivates human trypsinogen, whereas cathepsin L-deletion reduces the severity of pancreatitis in mice. Gastroenterology 138(2): 726-737, 2010. PMID: 19900452.
- Whitcomb DC,Gorry MC,Preston RA,Furey W,Sossenheimer MJ,Ulrich CD, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet 14(2): 141-145, 1996. PMID: 8841182.