
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2013.21
| Attachment | Size |
|---|---|
| 527.04 KB |
Introduction
Autoimmune pancreatitis (AIP) is characterized by pancreatic swelling and irregular narrowing of the main pancreatic duct, which often mimic pancreatic cancer (1, 17, 18, 20). AIP was recently classified into two types based on the pathological differences: type 1 for lymphoplasmacytic sclerosing pancreatitis (LPSP) and type 2 for idiopathic duct centric chronic pancreatitis (IDCP) or AIP with granulocytic epithelial lesion (GEL) (39). Type 1 AIP is closely associated with increased IgG4 antibodies in the serum and affected tissues (6, 7, 39, 47). Although the long-term prognosis and outcomes are relatively well described in type 1 AIP, these outcomes are less understood in type 2 AIP. Accordingly the following discussion primarily refers to type 1 AIP, while what is understood regarding type 2, which is almost exclusively found in Western countries, is discussed at the end (8, 28).
Long-term prognosis and outcome of type 1 AIP
Most patients with type 1 AIP (referred to as “AIP” in this section) respond favorably to corticosteroid therapy, which results in the amelioration of symptomatic, radiographic, serologic, and pathologic findings. It is possible for patients to have a spontaneous recovery. However, during long-term follow-up, some patients with AIP are noted to progress to the advanced stage of pancreatic stone formation after recurrence, which may be similar to the findings of chronic pancreatitis (Figure 1) (29, 30, 40). In addition, the possibility of an association with malignant conditions, such as pancreatic cancer or other malignancies has been reported (4, 5, 10, 32, 37, 44).
Progression to chronic pancreatitis
AIP is characterized by high serum IgG4 concentration,IgG4-positive staining plasma cell infiltration in affected pancreatic tissue, and a favorable response to corticosteroid therapy. Imaging analyses by ultrasonography (US), computed tomography (CT), and endoscopic retrograde cholangiopancreatography (ERCP) show sonolucent (i.e., hypoechoic) swelling and irregular narrowing of the main pancreatic duct, both of which are due to lymphoplasmacytic inflammation at the acute stage. In 1995, Yoshida et al. first proposed the concept of AIP, which was considered to be free from calcification and to rarely progress to ordinary chronic pancreatitis (46). Although most patients have a favorable response to corticosteroid therapy, some develop pancreatic atrophy and stone formation with irregular dilatation of the main pancreatic duct (MPD) (30, 40). These imaging findings mimic those of chronic pancreatitis, suggesting that in some cases AIP may progress into chronic pancreatitis.
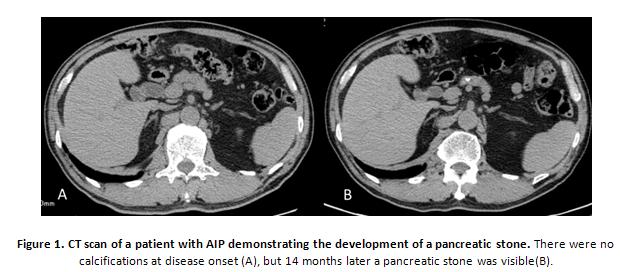
If this is the case, ordinary chronic pancreatitis could also include the advanced stage of AIP. This is supported by the observation that serum IgG4 remains elevated in over 60% of patients after clinical improvement (19). To clarify whether ordinary chronic pancreatitis includes the advanced stage of AIP, we measured serum levels of IgG4 in 175 patients with chronic pancreatitis who had been diagnosed before 1995, when the concept of AIP was first proposed. High serum IgG4 concentrations were found in 7.4% of patients with ordinary chronic pancreatitis, suggesting that the advanced stage of AIP may result in the development of ordinary chronic pancreatitis (21). Similarly, serum IgG4 was elevated in 11.9% of sera from Korean patients with ordinary chronic pancreatitis (2). A French study showed that more than one third of AIP patients developed pancreatic imaging abnormalities of atrophy, calcification, and/or duct irregularities and functional insufficiency within 3 years of diagnosis (28). Finally, one autopsy case of AIP showed similar pathological findings to chronic pancreatitis instead of the typical AIP findings of abundant lymphoplasmacytic infiltration, IgG4-bearing plasma cell infiltration, and obstructive phlebitis (25).
Pancreatic stone formation
Features of chronic pancreatitis include clinical findings of exocrine or endocrine dysfunction, imaging findings of pancreatic calcifications in the parenchyma or duct, and irregular MPD dilatation, and pathological findings of acinar or ductal cell loss, fibrosis, and stone formation. Of all these findings, pancreatic stone formation is a representative imaging finding that particularly correlates well with functional and pathological abnormalities.
The reported prevalence of pancreatic stone formation in AIP has been variable. Increased or de novo stone formation, including small calculi, was seen in 28 of 69 (41%) patients followed for at least 3 years at our institution (Shinshu University Hospital). Multivariate analysis identified narrowing of both Wirsung’s and Santorini’s ducts at diagnosis as an independent risk factor for pancreatic stone formation, which presumably led to pancreatic juice stasis and stone development (30). A long-term follow up study showed 16 of 73 (22%) AIP patients progressed to chronic pancreatitis that fulfilled the revised Japanese clinical diagnostic criteria for chronic pancreatitis in the chronic stage (29). However, other studies have indicated a lower prevalence of pancreatic stone formation during long term follow-up (41, 42). A recent multicenter, international analysis estimated that pancreatic stones occurred in only 7% of subjects with follow-up imaging permitting evaluation for stone disease (8). Further studies are needed to explain these discrepancies, and to understand if the formation of stones can be prevented.
Disease relapse
AIP is a chronic disease that can have a relapsing clinical course. To illustrate the frequency and distribution of disease relapses we reviewed the medical charts of 84 patients with AIP, who were followed up for more than 1 year at Shinshu University Hospital. Twenty-eight of the 84 patients (33%) experienced a total of 60 recurrences, including autoimmune pancreatitis (n = 26 times), sclerosing cholangitis (n = 18), lacrimal and salivary gland lesions (n = 5), and retroperitoneal fibrosis (n = 4). Seventy-two percent of the recurrences occurred in the maintenance stage of corticosteroid therapy. Although no markers at diagnosis significantly predicted recurrence, IgG and immune complexes tended to be elevated in the relapse group compared to the non-relapse group. During clinical follow-up, the development of pancreatic stones was more frequent in the relapse group (14 patients, 50%) than in the non-relapse group (13 patients, 23%). Collectively, one-third of patients with AIP developed a pancreatic stone. Close observation with activity markers during follow-up and early intervention with corticosteroid therapy may help to prevent recurrence in such cases (22).
Published series have reported similar relapse rates in AIP ranging from 30% to 50% (9, 16, 24, 26, 40, 43). Patients with relapse generally experienced 1 or 2 episodes, although some experienced many relapses. Corticosteroid therapy was reported to significantly increase the remission rate and reduce the relapse rate of AIP (16, 26). Thus, corticosteroid therapy is currently considered the standard treatment for inducing remission in AIP (13). Although spontaneous remission occurs in some patients with AIP, these patients are usually good candidates for corticosteroid therapy (9, 13, 16, 24, 26). According to the Japanese Consensus Guidelines for Management of AIP, the indications for corticosteroid therapy in AIP patients are symptoms such as obstructive jaundice, abdominal pain, and back pain, and the presence of symptomatic extrapancreatic lesions (15). In principle, corticosteroid therapy should be administered for all patients diagnosed with AIP (15).
Since AIP is the pancreatic manifestation of IgG4-Related Disease, other manifestations of IgG4-related disease can be seen at disease relapse (23, 38). In addition to pancreatic lesions, other common manifestations include sclerosing cholangitis, lacrimal/salivary gland lesions, retroperitoneal fibrosis, and interstitial pneumonitis (8, 24). These lesions also respond well to corticosteroid therapy. In our study population, the first, second, and third recurrences occurred at medians of 33, 66, and 122 months following steroid therapy, and 72% of recurrences occurred during the maintenance therapy stage. Other studies have shown that relapse generally occurs within the first 3 years following diagnosis (26). In those who develop a relapse, 56% relapsed within 1 year, and 92% relapsed within 3 years from the start of steroid treatment (16). Although relapse in our study occurred mostly during the maintenance stage of corticosteroid therapy, the relapse rate of patients with AIP on maintenance treatment was 23%, which was significantly lower than patients who stopped maintenance treatment (34%) (16). According to the Japanese Consensus Guidelines for Management of AIP, maintenance therapy (2.5 – 5 mg/day) is recommended to prevent recurrence, and stopping of maintenance therapy should be planned within at least 3 years in cases with serologic and radiologic improvement (15).
Previous studies indicated that various factors at diagnosis, including involvement of proximal biliary tract, diffuse pancreatic swelling, jaundice, IgG4, immune complex, soluble IL2 receptor, and complement are predictive factors of relapse (9, 21, 24, 26, 41). Specific HLA antigens were reported to predict the recurrence of AIP, and substitution of aspartic acid at position 57 of HLA DQβ1 was reported to affect the recurrence of autoimmune pancreatitis (35). We reported that serum elevation of IgG4 and immune complex preceded the clinical manifestations of recurrence (18). Accordingly, serial measurements of IgG, IgG4, and immune complex in the follow-up period may be useful to predict recurrence (13, 18, 21).
Relapse after surgical resection of the pancreas
Recently, Detlefsen et al. reported that 21 of 51 AIP patients (41.2%) who underwent surgical resection of the pancreas experienced recurrence during long term follow-up; the sites of recurrence were the pancreas (n = 8) and extrapancreatic bile ducts (n = 7) (3). Recurrence rate and sites were similar to those of the non-resection group. Their results are in contrast to a previous study, which showed a decreased risk of relapse in those undergoing surgical resection (36).
Pancreatic function
Pancreatic exocrine function
AIP is associated with exocrine dysfunction in 83% – 88% of cases during the acute inflammatory stage (11, 13, 14, 34). Following corticosteroid treatment and during the chronic stage, exocrine dysfunction resolves in most patients. However, exocrine dysfunction persists or may develop during long-term follow-up in some patients, which may be associated with the transition to chronic pancreatitis (42).
Pancreatic endocrine function
Diabetes mellitus occurs in 42% – 78% of cases during the acute stage of AIP (11, 13, 14, 33, 34). Similar to exocrine dysfunction, endocrine dysfunction, especially diabetes mellitus, is often ameliorated after corticosteroid therapy (9, 31, 34, 42). Miyamoto et al. reported amelioration of diabetes mellitus in 10 of 16 (63%) AIP patients 3 years after corticosteroid therapy, indicating that corticosteroid therapy is often effective for the treatment of diabetes in AIP (31). However, corticosteroid therapy sometimes causes deterioration of glycemic control, especially in aged patients, and thus requires cautious administration (33). Ito et al. reported that 10 of 50 AIP patients who received insulin treatment experienced hypoglycemic attacks, suggesting the need for vigilance when insulin therapy is administered (12). One third of AIP patients with diabetes mellitus suffered from diabetes at the onset of AIP; these patients frequently had a family history of diabetes mellitus and had poor nutritional status. Half of AIP patients are diagnosed with diabetes mellitus at AIP onset, however only 10% of AIP patients continued to have diabetes mellitus after corticosteroid therapy (12, 33).
AIP and complications of pancreatic cancer and other malignancies
Chronic pancreatitis has been regarded as risk factor for the occurrence of pancreatic cancer (27). Therefore, if AIP can progress to chronic pancreatitis, it also may be complicated with pancreatic cancer. A Japanese survey indicated that the average life expectancies of male and female patients with chronic pancreatitis were 11 and 17 years shorter than those of the general population, respectively. The major cause of the death was malignancy, indicating that the standard death rates for bile duct and pancreatic cancer were very high (3.44 and 7.84, respectively). It is possible that immunodeficiency due to corticosteroid therapy and chronic inflammation of the pancreas may contribute to the occurrence of malignancy.
There have been a few previous reports of AIP complicated with pancreatic cancer (4, 5, 10, 32, 44). Characteristic features of pancreatic cancer complicated with AIP are more frequent occurrence at body and tail regions compared with ordinary pancreatic cancer,(15) and earlier occurrence after the diagnosis of AIP compared with chronic pancreatitis. These results raise the possibility that AIP may contribute to the occurrence of pancreatic cancer, however these cases are highly subject to selection bias.
Since AIP occurs predominantly in elderly patients, deficiency of the immunosurveillance system may be associated with its pathogenesis, which in turn may be associated with the occurrence of various malignancies other than pancreatic cancer (37). In addition to AIP, IgG4-related disease was reported to be highly complicated with malignancies (45). In clinical follow-up for AIP and IgG4-related disease, caution is recommended to monitor for the occurrence of malignancy, however further studies are needed to clarify the true risk and most appropriate methods of cancer surveillance.
Long-term prognosis and outcome of type 2 AIP
The long-term prognosis and outcome of type 2 AIP have not been fully clarified. The two subtypes can be definitively distinguished based on their histology (See Histology of Autoimmune Pancreatitis). Type 2 AIP patients are younger than those with type 1 AIP, do not show the male gender bias seen in type 1 AIP, and are unlikely to have elevation of serum IgG4 or other organ involvement (39). A multicenter international analysis showed that the average ages at diagnosis were 61.4 and 39.9 years for type 1 and type 2 AIP, respectively, and the proportion of males was 77% in type 1 and 55% in type 2 AIP. In addition, type 2 AIP represented a smaller proportion of AIP in Asian countries compared with European and North American countries (8).
During the acute stage, imaging findings of type 2 AIP appear similar to those of type 1, including pancreatic swelling and irregular narrowing of the MPD. Similar to type 1 AIP, those with type 2 AIP respond favorably to corticosteroid therapy. However, the recurrence rate of type 2 AIP was significantly lower than that of type 1, and the site of type 2 AIP recurrence was limited to the pancreas. Few pancreatic stones were found in type 2 AIP during follow-up, suggesting it is uncommon for type 2 AIP to progresses to an advanced stage (8). However, another study indicated that the outcome of patients with type 2 AIP was not different from that of patients with type 1 AIP, except for diabetes, which was significantly higher in type 1 AIP (28). Further studies are therefore needed to better define the long-term prognosis and outcomes of type 2 AIP.
Summary
Type 1 AIP is a chronic, relapsing disease. Although the acute inflammatory phase is very responsive to corticosteroid therapy, there are several potential long term complications that can develop. Endocrine and exocrine pancreatic dysfunction is more typical during the acute phase. They may resolve with corticosteroid therapy, but occur later when the pancreas has atrophied. Disease relapses are common and can develop in the pancreas, biliary tree, or other distant sites associated with IgG4-RD. Careful observation of prodromal symptoms and activity markers during follow-up, as well as early intervention with corticosteroid therapy may help to limit morbidity from disease relapses. Pancreatic duct stones can develop, and are more likely in those with relapsing disease. There is a theoretical increased risk for developing pancreatic cancer, but the actual risk is not fully understood. In contrast, in type 2 AIP disease relapse and other long term complications are uncommon.
Acknowledgements
We would like to thank Drs. Takashi Muraki, Tetsuya Ito, Keita Kanai, Takaya Oguchi, Hideaki Hamano, and Norikazu Arakura for their clinical assistance and contributions to this work.
References
- Chari ST, Takahashi N, Levy MJ, Smyrk TC, Clain JE, Pearson RK, Petersen BT, Topazian MA, and Vege SS. A diagnostic strategy to distinguish autoimmune pancreatitis from pancreatic cancer. Clin Gastroenterol Hepatol 7: 1097-1103, 2009. PMID: 194100172
- Choi EK, Kim MH, Lee TY, Kwon S, Oh HC, Hwang CY, Seo DW, Lee SS, and Lee SK. The sensitivity and specificity of serum immunoglobulin G and immunoglobulin G4 levels in the diagnosis of autoimmune chronic pancreatitis: Korean experience. Pancreas 35: 156-161, 2007. PMID: 17632322
- Detlefsen S, Zamboni G, Frulloni L, Feyerabend B, Braun F, Gerke O, Schlitter AM, Esposito I, and Kloppel G. Clinical features and relapse rates after surgery in type 1 autoimmune pancreatitis differ from type 2: a study of 114 surgically treated European patients. Pancreatology 12: 276-283, 2012. PMID: 22687385
- Fukui T, Mitsuyama T, Takaoka M, Uchida K, Matsushita M, and Okazaki K. Pancreatic cancer associated with autoimmune pancreatitis in remission. Intern Med 47: 151-155, 2008. PMID: 18239323
- Ghazale A and Chari S. Is autoimmune pancreatitis a risk factor for pancreatic cancer? Pancreas 35: 376, 2007. PMID: 18090248
- Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, Fukushima M, Nikaido T, Nakayama K, Usuda N, and Kiyosawa K. High serum IgG4 concentrations in patients with sclerosing pancreatitis. New Engl J Med 344: 732-738, 2001. PMID: 11236777
- Hamano H, Kawa S, Ochi Y, Unno H, Shiba N, Wajiki M, Nakazawa K, Shimojo H, and Kiyosawa K. Hydronephrosis associated with retroperitoneal fibrosis and sclerosing pancreatitis. Lancet 359: 1403-1404, 2002. PMID: 11978339
- Hart PA, Kamisawa T, Brugge WR, Chung JB, Culver EL, Czako L, Frulloni L, Go VL, Gress TM, Kim MH, Kawa S, Lee KT, Lerch MM, Liao WC, Lohr M, Okazaki K, Ryu JK, Schleinitz N, Shimizu K, Shimosegawa T, Soetikno R, Webster G, Yadav D, Zen Y, and Chari ST. Long-term outcomes of autoimmune pancreatitis: a multicentre, international analysis. Gut, 2012. PMID: 23232048
- Hirano K, Tada M, Isayama H, Yagioka H, Sasaki T, Kogure H, Nakai Y, Sasahira N, Tsujino T, Yoshida H, Kawabe T, and Omata M. Long-term prognosis of autoimmune pancreatitis with and without corticosteroid treatment. Gut 56: 1719-1724, 2007. PMID: 17525092
- Inoue H, Miyatani H, Sawada Y, and Yoshida Y. A case of pancreas cancer with autoimmune pancreatitis. Pancreas 33: 208-209, 2006. PMID: 16868495
- Ito T, Kawabe K, Arita Y, Hisano T, Igarashi H, Funakoshi A, Sumii T, Yamanaka T, and Takayanagi R. Evaluation of pancreatic endocrine and exocrine function in patients with autoimmune pancreatitis. Pancreas 34: 254-259, 2007. PMID: 17312466
- Ito T, Nakamura T, Fujimori N, Niina Y, Igarashi H, Oono T, Uchida M, Kawabe K, Takayanagi R, Nishimori I, Otsuki M, and Shimosegawa T. Characteristics of pancreatic diabetes in patients with autoimmune pancreatitis. Journal of digestive diseases 12: 210-216, 2011. PMID: 21615876
- Ito T, Nishimori I, Inoue N, Kawabe K, Gibo J, Arita Y, Okazaki K, Takayanagi R, and Otsuki M. Treatment for autoimmune pancreatitis: consensus on the treatment for patients with autoimmune pancreatitis in Japan. J Gastroenterol 42 Suppl 18: 50-58, 2007. PMID: 17520224
- Kamisawa T, Egawa N, Inokuma S, Tsuruta K, Okamoto A, Kamata N, Nakamura T, and Matsukawa M. Pancreatic endocrine and exocrine function and salivary gland function in autoimmune pancreatitis before and after steroid therapy. Pancreas 27: 235-238, 2003. PMID: 14508128
- Kamisawa T, Okazaki K, Kawa S, Shimosegawa T, and Tanaka M. Japanese consensus guidelines for management of autoimmune pancreatitis: III. Treatment and prognosis of AIP. J Gastroenterol 45: 471-477, 2010. PMID: 20213336
- Kamisawa T, Shimosegawa T, Okazaki K, Nishino T, Watanabe H, Kanno A, Okumura F, Nishikawa T, Kobayashi K, Ichiya T, Takatori H, Yamakita K, Kubota K, Hamano H, Okamura K, Hirano K, Ito T, Ko SB, and Omata M. Standard steroid treatment for autoimmune pancreatitis. Gut 58: 1504-1507, 2009. PMID: 19398440
- Kawa S, Fujinaga Y, Ota M, Hamano H, and Bahram S. Autoimmune Pancreatitis and Diagnostic Criteria. Current Immunology Reviews 7: 144-161, 2011.
- Kawa S and Hamano H. Clinical features of autoimmune pancreatitis. J Gastroenterol 42 Suppl 18: 9-14, 2007. PMID: 17520217
- Kawa S, Hamano H, and Kiyosawa K. High serum IgG4 concentrations in patients with sclerosing pancreatitis. Reply. New Engl J Med 345: 148-148, 2001. PMID: 11236777
- Kawa S, Hamano H, and Kiyosawa K. Pancreatitis. In: The Autoimmune Diseases (4th ed.), edited by Rose N and MacKay I. St Louis: Academic Press, 2006, p. 779-786.
- Kawa S, Hamano H, Ozaki Y, Ito T, Kodama R, Chou Y, Takayama M, and Arakura N. Long-term follow-up of autoimmune pancreatitis: characteristics of chronic disease and recurrence. Clin Gastroenterol Hepatol 7: S18-22, 2009. PMID: 19896092
- Kawa S, Ito T, Watanabe T, Maruyama M, Yoneda S, Maruyama M, Kodama R, Ozaki Y, Muraki T, Hamano H, and Arakura N. Frequency and prevention of autoimmune pancreatitis relapse in a Japanese population. 4th AOPA & KPBA, Jeju, 2011, p. 27-33.
- Kawa S and Sugai S. History of Autoimmune Pancreatitis and Mikulicz's Disease. Current Immunology Reviews 7: 137-143 2011.
- Kim HM, Chung MJ, and Chung JB. Remission and relapse of autoimmune pancreatitis: focusing on corticosteroid treatment. Pancreas 39: 555-560, 2010. PMID: 20182397
- Kitano Y, Matsumoto K, Chisaka K, Imazawa M, Takahashi K, Nakade Y, Okada M, Aso K, Yokoyama K, Yamamoto M, Yoshie M, Ogawa K, and Haneda M. An autopsy case of autoimmune pancreatitis. JOP 8: 621-627, 2007. PMID: 17873471
- Kubota K, Watanabe S, Uchiyama T, Kato S, Sekino Y, Suzuki K, Mawatari H, Iida H, Endo H, Fujita K, Yoneda M, Takahashi H, Kirikoshi H, Kobayashi N, Saito S, Sugimori K, Hisatomi K, Matsuhashi N, Sato H, Tanida E, Sakaguchi T, Fujisawa N, and Nakajima A. Factors predictive of relapse and spontaneous remission of autoimmune pancreatitis patients treated/not treated with corticosteroids. J Gastroenterol, 2011. PMID: 21491208
- Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, Dimagno EP, Andren-Sandberg A, and Domellof L. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. New England J Med 328: 1433-1437, 1993. PMID: 8479461
- Maire F, Le Baleur Y, Rebours V, Vullierme MP, Couvelard A, Voitot H, Sauvanet A, Hentic O, Levy P, Ruszniewski P, and Hammel P. Outcome of Patients With Type 1 or 2 Autoimmune Pancreatitis. Am J Gastroenterol, 2010. PMID: 20736934
- Maruyama M, Arakura N, Ozaki Y, Watanabe T, Ito T, Yoneda S, Maruyama M, Muraki T, Hamano H, Matsumoto A, Kawa S. Type 1 Autoimmune Pancreatitis Can Transform into Chronic Pancreatitis: A Long-Term Follow-Up Study of 73 Japanese Patients. Int Rheumatol 2013: 8, 2013. PMID: 23762066
- Maruyama M, Arakura N, Ozaki Y, Watanabe T, Ito T, Yoneda S, Muraki T, Hamano H, Matsumoto A, and Kawa S. Risk factors for pancreatic stone formation in autoimmune pancreatitis over a long-term course. J Gastroenterol, 2011. PMID: 22183858
- Miyamoto Y, Kamisawa T, Tabata T, Hara S, Kuruma S, Chiba K, Inaba Y, Kuwata G, Fujiwara T, Egashira H, Koizumi K, Sekiya R, Fujiwara J, Arakawa T, Momma K, and Asano T. Short and long-term outcomes of diabetes mellitus in patients with autoimmune pancreatitis after steroid therapy. Gut and liver 6: 501-504, 2012. PMID: 23170157
- Motosugi U, Ichikawa T, Yamaguchi H, Nakazawa T, Katoh R, Itakura J, Fujii H, Sato T, Araki T, and Shimizu M. Small invasive ductal adenocarcinoma of the pancreas associated with lymphoplasmacytic sclerosing pancreatitis. Pathol Int 59: 744-747, 2009. PMID: 19788620
- Nishimori I, Tamakoshi A, Kawa S, Tanaka S, Takeuchi K, Kamisawa T, Saisho H, Hirano K, Okamura K, Yanagawa N, and Otsuki M. Influence of steroid therapy on the course of diabetes mellitus in patients with autoimmune pancreatitis: findings from a nationwide survey in Japan. Pancreas 32: 244-248, 2006. PMID: 16628078
- Nishino T, Toki F, Oyama H, Shimizu K, and Shiratori K. Long-term outcome of autoimmune pancreatitis after oral prednisolone therapy. Intern Med 45: 497-501, 2006. PMID: 16702740
- Park do H, Kim MH, Oh HB, Kwon OJ, Choi YJ, Lee SS, Lee TY, Seo DW, and Lee SK. Substitution of aspartic acid at position 57 of the DQbeta1 affects relapse of autoimmune pancreatitis. Gastroenterology 134: 440-446, 2008. PMID: 18155707
- Sah RP, Chari ST, Pannala R, Sugumar A, Clain JE, Levy MJ, Pearson RK, Smyrk TC, Petersen BT, Topazian MD, Takahashi N, Farnell MB, and Vege SS. Differences in Clinical Profile and Relapse Rate of Type 1 vs Type 2 Autoimmune Pancreatitis. Gastroenterology, 2010. PMID: 20353791
- Shiokawa M, Kodama Y, Yoshimura K, Kawanami C, Mimura J, Yamashita Y, Asada M, Kikuyama M, Okabe Y, Inokuma T, Ohana M, Kokuryu H, Takeda K, Tsuji Y, Minami R, Sakuma Y, Kuriyama K, Ota Y, Tanabe W, Maruno T, Kurita A, Sawai Y, Uza N, Watanabe T, Haga H, and Chiba T. Risk of cancer in patients with autoimmune pancreatitis. Am J Gastroenterol 108: 610-617, 2013. PMID: 23318486
- Stone JH, Zen Y, and Deshpande V. IgG4-related disease. The New England journal of medicine 366: 539-551, 2012. PMID: 22316447
- Sugumar A, Kloppel G, and Chari ST. Autoimmune pancreatitis: pathologic subtypes and their implications for its diagnosis. Am J Gastroenterol 104: 2308-2310, 2009. PMID: 19727085
- Takayama M, Hamano H, Ochi Y, Saegusa H, Komatsu K, Muraki T, Arakura N, Imai Y, Hasebe O, and Kawa S. Recurrent attacks of autoimmune pancreatitis result in pancreatic stone formation. Am J Gastroenterol 99: 932-937, 2004. PMID: 15128363
- Takuma K, Kamisawa T, Tabata T, Inaba Y, Egawa N, and Igarashi Y. Short-term and long-term outcomes of autoimmune pancreatitis. Eur J Gastroenterol Hepatol 23: 146-152, 2011. PMID: 21287714
- Uchida K, Yazumi S, Nishio A, Kusuda T, Koyabu M, Fukata M, Miyoshi H, Sakaguchi Y, Fukui T, Matsushita M, Takaoka M, and Okazaki K. Long-term outcome of autoimmune pancreatitis. J Gastroenterol 44: 726-732, 2009. PMID: 19396390
- Wakabayashi T, Kawaura Y, Satomura Y, Watanabe H, Motoo Y, and Sawabu N. Long-term prognosis of duct-narrowing chronic pancreatitis: strategy for steroid treatment. Pancreas 30: 31-39, 2005. PMID: 15632697
- Witkiewicz AK, Kennedy EP, Kennyon L, Yeo CJ, and Hruban RH. Synchronous autoimmune pancreatitis and infiltrating pancreatic ductal adenocarcinoma: case report and review of the literature. Hum Pathol 39: 1548-1551, 2008. PMID: 18619645
- Yamamoto M, Takahashi H, Tabeya T, Suzuki C, Naishiro Y, Ishigami K, Yajima H, Shimizu Y, Obara M, Yamamoto H, Himi T, Imai K, and Shinomura Y. Risk of malignancies in IgG4-related disease. Modern rheumatology / the Japan Rheumatism Association 22: 414-418, 2012. PMID: 21894525
- Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, and Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci 40: 1561-1568, 1995. PMID: 7628283
- Zen Y, Bogdanos DP, and Kawa S. Type 1 autoimmune pancreatitis. Orphanet J Rare Dis 6: 82, 2011. PMID: 22151922