
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2015.13
| Attachment | Size |
|---|---|
| 762.44 KB |
1. Introduction
Severe, disabling abdominal pain is the hallmark of chronic pancreatitis. Currently available treatments for pancreatitis pain are inadequate and expensive, both in health care dollars and in lost productivity. Pain is the most common reason for hospitalization among chronic pancreatitis patients, and as many as 40% require 3 or more admissions during their lifetime for pain management (72). Improved treatments depend on a better understanding of the mechanisms of chronic visceral pain, a subject that has gained attention recently with the development of suitable animal models and reproducible experimental measures of sustained pancreatic pain.
2. Manifestations and Treatment of Pancreatic Pain
Pain Theories
Traditional theories of the origin of pancreatic pain in chronic pancreatitis focused on structural abnormalities causing ductal hypertension (11). Such abnormalities ranged from stones and strictures, to fibrosis due to toxic effects and ischemia (27). While this ductal obstruction theory is logical, studies in patients with chronic pancreatitis have failed to show a correlation between ductal pressure and pain levels; moreover, ductal pressures do not accurately predict the success of ductal decompression procedures (34-37). In fact, Bornman et al demonstrated that there was no significant difference in either the anatomy or the morphological changes between groups of patients with either painful or painless pancreatitis (10). Rather than a single mechanism of pain, recent research has favored a more complex relationship between these structural and morphological components, and their interaction with neurobiological mechanisms (33). Nociceptive pathways, inflammatory mediators and sensitization of both central and peripheral pathways have been shown to play important roles in pancreatic pain (76).
Chronic Pain Syndrome: A downward spiral
Among the many clinical sequelae of chronic pancreatitis, pain has been shown to be the most important factor affecting quality of life (78). The pain often becomes the focal point around which work, leisure activities and relationships must revolve. Two types of pain patterns have been identified among these patients, type A, characterized by intermittent flares of pain, and type B, consisting of prolonged periods of persistent pain of varying severity (3). In the largest study of pain in chronic pancreatitis, Mullady et al showed that those who exhibit more type B pain of a more constant nature, have lower quality of life measures (72). In a study of 265 patients, Wehler et al showed that as abdominal pain index scores increased across subgroups, there was a significant and profound decrease in all quality of life indices. Since eating can trigger pain exacerbations, patients typically respond by decreasing food intake. Many patients also suffer nutrient malabsorption due to pancreatic exocrine insufficiency, and this combination leads to progressive weight loss and malnutrition. Decreased BMI has been correlated with impairment in quality of life measurements (102).
Medical Treatment
The mainstay therapy for chronic pancreatitis is the symptomatic treatment of pain. Since we have a limited understanding of the pathogenesis of chronic pancreatic pain, treatment is limited to a supportive care regimen targeting symptoms rather than etiologies of the pain (40). Therapeutic regimens rely heavily on opioid analgesics, which lead to both physiological and psychological dependence, as well as tolerance requiring escalating doses. The undesirable side effects of these drugs reduce patient well-being through physical symptoms such as somnolence, impaired cognitive function, and constipation. The side effects of existing therapeutics combine with inadequately treated pain to produce the detrimental socioeconomic effects of inability to work or, in some cases, even to leave the house (83). Some studies have even supported the possible role of long term opioid treatment in the development of hyperalgesia and allodynia which further exacerbate pain syndromes (66). Newer approaches typically use multimodality combinations of agents that target inflammation, nerve injury, and descending pathways, with the goal of reducing narcotic dosages and achieving synergistic effects. Examples of this include the addition of the gabapentoid pregabalin, as well as the antioxidant methionine, both of which have been shown to improve pain in chronic pancreatitis (75, 90). Despite these advances, the ideal medical treatment remains elusive due to the lack of reliable trials comparing various treatment regimens, as well as the heterogeneity in pain patterns (5, 40).
Interventional Treatment
For pain that is refractory to medical management, an array of procedures have been used with risk and side effect profiles that roughly parallel their efficacy and durability in improving pain.
Peripheral nerve ablation
One of the original approaches was endoscopic injection of anti-inflammatory, analgesic or ablative agents into the celiac ganglia through which most of the pancreatic afferents pass (25). In a prospective study by Gress and colleagues, of 90 patients with chronic pancreatitis, only 55% of patients had significant improvement in pain scores following EUS-guided celiac plexus block, and only 10% had lasting benefit from the procedure at 24 weeks (47). Rare but serious side effects have included motor nerve impairment and exacerbation of pain. Therefore, it is no longer recommended as a routine therapy for patients with intractable benign pancreatic pain, but is still used to improve quality of remaining life for some patients with severe pain from pancreatic cancer (71). Newer therapies have been developed utilizing percutaneous radiofrequency ablation to achieve site directed ablation of splanchnic nerves. Demonstrated first in a small cohorts of patients with chronic non-malignant abdominal pain by Garcea et al in 2005 (41), this approach was used by Verhaegh et al in 2013 on a cohort of 11 chronic pancreatitis patients. They found a 50-75% reduction in pain scores in greater than half of patients, and a median pain free period of 45 weeks (98). While these early results have been promising, the durability of the effect remains a limitation, and evidence for success of repeat interventions is lacking. Surgical resection of segments of these peripheral splanchnic nerves via a minimally invasive approach, so called thoracoscopic splanchnicectomy, has also been shown to be effective for short-term pain relief, but durable improvement remains elusive for 50% of patients after 15 months (4). For these reasons, peripheral nerve interventions play a limited role in the clinical management of patients with severe chronic pancreatitis pain. They may, for example, be useful as a bridge therapy to reduce narcotic dependence, and, in suitable cases, may allow a patient to gain weight in preparation for a more durable, and higher risk, surgical procedure.
Pancreatic Drainage
For a select minority of patients with diffuse dilation of the main pancreatic duct, typically due to anatomic, fibrotic, or calculus obstruction at or near the insertion of the pancreatic duct into the common channel, endoscopic or surgical drainage into the small intestine may provide benefit. Endoscopically, this is typically achieved via a transampullary approach during endoscopic retrograde cholangiopancreatography (ERCP) with sphincterotomy, dilation and possibly stenting (95). For patients with large duct disease, decompression can improve pain, however, repeat endoscopic therapy is often needed as more than half of patients will have recurrence of pain (24). Extracorporeal shockwave lithotripsy (ESWL) can also be used as an adjunct with ERCP in patients with large duct stones (91). Surgical drainage procedures have evolved over time and the modern lateral pancreaticojejunostomy now typically includes resection of a portion of the pancreatic head (Frey procedure) to provide wide open drainage of pancreatic juice into the limb of jejunum. One study of 29 chronic pancreatitis patients showed long term pain relief in 90% at one year when treated with the Frey procedure (95). Only two large randomized control trials have been done to date comparing surgical drainage with endoscopic drainage modalities. Both studies demonstrated significantly better long-term pain relief in surgically treated patients. Dite et al demonstrated that 37% of patients in the surgical group remained pain-free at their 5 year follow up (12). There were no significant differences in the number of adverse events between the two cohorts (12, 32).
Pancreatic Resection
For the majority of patients with debilitating chronic pancreatitis pain, the main pancreatic duct is not diffusely dilated, and drainage is not feasible. These patients with so called “small duct” disease that fail non-surgical management due to uncontrolled pain and its sequelae, and/or intolerance of the side effects of high dose narcotics, are relegated to the last ditch surgical alternative, that of resection. In general, the likelihood of pain relief and of surgical diabetes both scale with the percentage of pancreas removed, with more recent long-term follow up data diminishing enthusiasm for resection. In a study of 224 patients, Riediger et al evaluated surgical partial resections for chronic pancreatitis including pylorus preserving pancreaticoduodenectomy (PPPD), duodenum preserving pancreatic head resection (DPPHR), classic Whipple, distal pancreatectomy and central pancreatic resection. With a median follow up of 56.3 months, 60% of patients remained pain free at last follow-up. Although subgroup analysis by resection type did not demonstrate a significant correlation with pain outcomes, patient selection likely plays an important role (82). An advantage of more aggressive resections such as total pancreatectomy may be the added resection of pancreatic nerve ganglia (25). With more generalizable results showing improved glycemic control among patients that undergo total pancreatectomy with islet cell harvest and auto transplantation (44, 100), this procedure has recently gained some favor, however, the refractory nature of pain in a substantial fraction of these patients, even following the resource-intensive removal of the entire gland, remains both confusing and frustrating (89). This group of patients provides us with an interesting glimpse into the complexity of the extra-pancreatic pathways that contribute to sustained pancreatic pain.
3. Models
Whereas pancreatic atrophy and fibrosis can be induced experimentally in a variety of ways, measures of visceral pain have proven more difficult. Studies in rats evaluated spontaneous activity qualitatively by video tracking and sensitivity of the abdomen to mechanical and electrical stimulation (105). The most widely used of these rat models was developed in 1996 by Puig et al who injected trinitrobenzene sulfonic acid (TNBS) directly into the pancreatic duct of rats to induce early severe acute pancreatitis that evolved over weeks into painful chronic pancreatitis (80). This model was further characterized and modified to provide better face validity and generalization to human disease by Winston et al in 2005 (105).This model has proved invaluable in providing insight into the complex nature of pain from chronic pancreatitis. Further progress in identifying specific pathways that might be therapeutic targets, however, was hampered by the lack of a murine model in which putative mediators could be genetically deleted.
Adaptation of the TNBS model to mice was fraught with early experimental failure related to the high mortality of severe acute pancreatitis in the physiologically fragile mice. Our laboratory adapted the model to mice by dramatically reducing the dose of TNBS and by providing perioperative fluid resuscitation during the first 24 hours (15). The resultant chronic pancreatitis is apparent after 1-2 weeks with severe fibrosis, monocyte infiltration, atrophy, and fatty replacement of the gland. We used Von Frey filament probing of the abdomen to demonstrate referred mechanical hyperalgesia, in which heightened withdrawal responses were measured to a mildly painful stimulus, as well as allodynia, in which probes that do not cause pain in control mice evoke withdrawal responses. TNBS-injected mice also showed reduced spontaneous activity (distance and time) on a running wheel, and longer periods of immobility during open field testing. This model can be used to examine both peripheral and central mechanisms of sustained pain and for comparison with models of somatic pain, such as peripheral or spinal nerve ligation, so that both shared and unique pathways can be identified.
4. Components of Pancreatic Pain
Nociceptive Neurons
In addition to parasympathetic cholinergic innervation from the vagus nerve, and sympathetic innervation mainly derived from the celiac ganglia, the pancreas is also innervated by nociceptive sensory neurons. These afferent neurons have their cell bodies in the dorsal root ganglia (DRG) and they give off projections that map to the dorsal horn of the spinal cord (76) (Figure 1). They are responsible for transmission of noxious visceral stimuli from the pancreas, and the relay of this information to the central nervous system.
Uncontrolled Proteolysis
The pancreas is rich in cysteine and serine proteases that can be released following a variety of insults and are known to activate, either directly or indirectly, nociceptive neurons. Using a near infrared-labeled activity-based probe that covalently modifies active cathepsins, our laboratory found significant accumulation of cathepsins B, L and S in both the inflamed rodent pancreas and in human juice from patients with painful chronic pancreatitis (63). Cathepsins, in turn, cleave and activate trypsinogens, yielding active trypsins, some of which are resistant to endogenous degradation by ubiquitous inhibitors, and are thereby free to bind and activate receptors on peptidergic neurons (14). Following activation, peptidergic neurons release neuropeptides and inflammatory mediators including calcitonin gene related peptide (CGRP), substance P (SP), vasoactive intestinal polypeptide (VIP) and bradykinin, that act both peripherally where they promote vasodilation, plasma extravasation, and neutrophil infiltration (so called neurogenic inflammation) and centrally where they activate central pain pathways (57).
Sensory Neuron Receptors
Vanilloid Receptors
One of the best characterized pain receptors is Transient Receptor Potential Vanilloid 1 (TRPV1). A member of the family of vanilloid nociceptive receptors found on sensory neurons, it functions as a non-selective cation channel, permitting flow of sodium and calcium into cells, leading to depolarization of the cell membrane and release of neurotransmitters such as SP and CGRP (76). Originally known as the capsaicin receptor, it is activated by heat and local acidification, as well as multiple endogenous chemical mediators including leukotrienes and arachadonic acid metabolites (53). Caterina et al used TRPV1 knockout mice to clearly demonstrate the role of TRPV1 in nociception and tissue-injury induced hyperalgesia (13). We showed that TRPV1 plays an important role in nociceptive mediation in acute pancreatitis through the induction of SP and CGRP release by pancreatic sensory nerves, increasing c-fos expression in the spinal cords of rats. Administration of a TRPV1 antagonist attenuated this effect (104). TRPV1 is upregulated in chronic pancreatitis and has been demonstrated as a mediator of hyperalgesia and inflammation in this condition (60). Additionally, it has been implicated to have interactions with other TRP receptors as well as protease activated receptor 2 (PAR2), a G-protein coupled receptor with unique roles in inflammation and pain sensitization (76), described below.
TRPV1 can work alone or in concert with other TRP receptors, such as TRP Ankyrin 1 (TRPA1), to amplify nociceptive signaling. Required for sensory neuron excitation, TRPA1 functions as a “gatekeeper” of chronic inflammation by serving two major roles: controlling peripheral release of inflammatory neuropeptides, and facilitating neuronal activation by inflammatory mediators released through local tissue injury (8). Though it had been previously shown to mediate inflammation and visceral pain in acute pancreatitis (17), the first evidence of TRPA1’s direct role in pain from chronic pancreatitis came in 2013 with the establishment of a TNBS murine model of chronic pancreatitis. In this model of painful chronic pancreatitis following severe acute pancreatitis, we found that TRPA1 knockout mice had less inflammation and fibrosis and markedly reduced pain indices, including referred mechanical hyperalgesia, spontaneous running activity, and mobility in open field testing, as compared with wild type controls (15).
Over the past decade, it has been demonstrated using knockout mice (9, 13, 56), TRPA1 knockdowns (74) and the use of antagonists (77), that TRPA1 works in concert with TRPV1 to mediate inflammation-induced stimulus transmission in sensory neurons. Evidence for a direct interaction between the two channels was shown by Staruschenko et al using FRET constructs of the respective channels (87). Recently, TRPA1 and TRPV1 were implicated in the transition from acute to chronic inflammation in the pancreas. Schwartz et al, using a cerulein model of acute pancreatitis, demonstrated that morphologic acute to chronic changes were mitigated by administration of TRP antagonists (84).
Increasing evidence also supports a role for TRPV4 in pancreatic pain. TRPV4 is directly activated by shear stress, osmotic stimuli and lipid mediators, as well as indirectly via G-protein coupled receptors that regulate TRP channels (73, 79). TRPV4 knockout mice (61) and TRPV4 knockdowns (2) have demonstrated abnormal osmotic regulation and decreased responses to changes in pressure and tonicity. Alessandri et al proposed the attractive notion that the “soup” of inflammatory mediators that surround local tissue injury, including bradykinin, SP, prostaglandin E2 (PGE2), serotonin and histamine, among others, may induce mechanical hyperalgesia through activation of TRPV4, sensitizing it for a triggering event. They demonstrated that activation of TRPV4 by hypotonic saline was enhanced in the presence of PGE2, and increased nociceptive behavior in rats. These effects were absent in their TRPV4 knockout rats (2). They also demonstrated the involvement of protein kinase A and C intracellular second messenger pathways in the activation of TRPV4 (1). This activation in turn mediates pain transmission through subsequent activation of nociceptive spinal neurons in the superficial laminae of the spinal cord. In the pancreas, we showed that injection of a TRPV4 agonist into the pancreatic duct increased c-Fos like immunoreactivity expression in the spinal cord in the input regions of pancreatic sensory neurons located by retrograde tracing, suggesting that TRPV4 could play a role in pain signaling in the inflamed pancreas (17). Further experiments are needed to clarify the importance of TRPV4 in acute and chronic pancreatic inflammatory pain.
Protease Activated Receptor 2 (PAR2)
Protease Activated Receptor 2 (PAR2) is one of four GPCRs that is activated by serine proteases such as trypsin and thrombin. These proteases cleave an N-terminal fragment, revealing a tethered receptor agonist (ligand), which can then bind and activate signaling pathways (99). Steinhoff et al provided initial evidence of a neurogenic inflammatory role for PAR2 by demonstrating its co-expression with neuropeptides CGRP and substance P in DRG neurons. Activation of PAR2 leads to release of neuropeptides in peripheral tissues as well as the spinal cord, increasing local inflammation and edema (88).
In addition to causing the direct release of inflammatory neuropeptides from sensory neurons, activated PAR2 leads to increased intracellular calcium, which lower the threshold for activation of TRP channels by other inflammatory mediators and products of tissue injury, so called “sensitization”. Thus, the addition of trypsin or PAR2 activating peptide (AcPep) in dorsal root ganglion cell culture led to significantly increased capsaicin - evoked CGRP release, an indication of PAR2 sensitization of TRPV1. In vivo, pre-injection of AcPep into the pancreatic duct increased capsaicin-induced FOS expression in pancreatic spinal cord segments compared with the control peptide, suggesting that PAR2 sensitizes TRPV1 in the pancreas (50). Under normal physiologic conditions, concentrations of active trypsin in the pancreas are low due to its release in a zymogen form as trypsinogen. However, following pancreatic inflammation, early activation of trypsins by cysteine proteases, as well as the recruitment of mast cells that release tryptase, can, in turn, activate PAR2 (28). Indirect evidence of the importance of mast cell products in chronic pancreatitis pain derives from the observation that mast cells were present in significantly higher numbers in patients with painful chronic pancreatitis than in patients with non-painful pancreatitis (33.8 vs. 9.4 average mast cell/10 high power field; p<0.01) or with healthy controls (33.8 vs. 6.1 average mast cell/10 high power field; p<0.01) (51). Intraductal injection of trypsin into the pancreatic duct of mice in sub-inflammatory concentrations, causes increased FOS expression in pancreatic specific spinal cord DRG. This effect was mitigated by pre-treatment with AcPep, indicating that PAR2 and trypsin may share this pain pathway (49). PAR2 activation in these neurons leads to sustained hyperalgesia (97). Thus, serine proteases contribute to pancreatic pain via multiple pathways mediated by PAR2 activation (14).
PAR2 has also been shown to sensitize both TRPV4 and TRPA1, and thereby lower the threshold for activation of pancreatic sensory neurons (26, 46). PAR2 – mediated sensitization of these TRP channels has been associated with neuropathic pain induced by the chemotherapy agent paclitaxel, which indicates that these pathways have clinical importance (19). Peripheral sensitization represents an important pathway by which the painful effects of inflammatory mediators that result from tissue injury are amplified and sustained.
Nerve Growth Factor (NGF) and receptor tyrosine kinase A (TrkA)
Nerve growth factor (NGF), a protein that contributes to the development and survival of neurons, also plays an important role in the peripheral sensitization of sensory neurons (106). It acts mainly through its high affinity tyrosine kinase receptor, TrkA, which is found in highest concentration within the pancreas in the perineurium. Co-expression of TrkA with NGF is increased in the pancreas from patients with chronic pancreatitis (39). NGF exerts its effects through multiple mechanisms including a direct effect on ion channels, post-translational modifications by second messengers, as well as by translocation of the NGF/trkA complex to the nucleus where it regulates transcriptional modifications to certain genes (107). Early evidence for its role in mediating visceral pain came from expression studies by McMahon et al in 1994, which demonstrated that almost all afferent neurons innervating visceral targets expressed trkA, while its expression in those innervating skeletal muscle was very low (68). Immunodepletion studies using a trkA-IgG on cultured neurons, showed a sustained hypoalgesia and a downregulation of CGRP (69). This was further supported by studies using animals lacking the trkA gene, which also experienced a significant hypoalgesic state (6). This same hypoalgesic effect was noted after administration of a blocking antibody for NGF to rats with chronic pancreatitis, which significantly increased A-type potassium currents, thereby decreasing the likelihood of depolarization (108). Conversely, both neonatal and adult rats that were injected with excess exogenous NGF showed a profound behavioral hyperalgesia (59). Recent reports suggest that NGF/trkA can sensitize neurons via interaction with the vanilloid receptor TRPV1, and NGF can regulate TRPV1 expression through both transcriptional and post-translational mechanisms (FIG 1) (107).
Neurokinin Receptor 1 (NK-1R)
Substance P, neurokinins A (NKA) and B (NKB) are the main tachykinins involved in sensory neural transmission and nociception. Substance P and NKA share a receptor, neurokinin receptor 1 (NK-1R), and NKB binds preferentially to neurokinin receptor 2 (NK-2R) (64). Through a study of human pancreatic tissue, Di Sabastiano et al found that although there was an increase in substance P surrounding pancreatic nerve fibers, there was not a concomitant increase in the gene encoding substance P. This observation led to the early understanding that substance P was being synthesized in extra-pancreatic ganglia and transported to the pancreas (29). Thus, activation of peripheral sensory nerve endings leads to the release of substance P and CGRP peripherally within the pancreas, where they promote neurogenic inflammation in a positive feedback loop that leads to amplification of inflammatory pain, and centrally, where substance P binds to NK-1R in the dorsal horn of the spinal cord and activates central pain pathways (45, 52). Shrikhande et al was the first to examine NK-1R expression in pancreata from patients with painful chronic pancreatitis. They established a definitive relationship between mRNA levels and intensity, frequency and duration of pain in these patients (85).
5. Central Sensitization
Nervous System Support Cells
Microglia
Microglia are immune cells in the central nervous system that respond to tissue injury by switching from a quiescent to an active state, in which they secrete inflammatory mediators to recruit other immune cells and promote cellular hypertrophy and proliferation (93, 103) Their function in the CNS is similar to that of macrophages in peripheral tissues.
How are microglia activated? The initial activation of microglia likely occurs through multiple pathways. Excitation of nociceptive neurons leads to release of the chemokine CCL2 that binds to its receptor CCR2 on microglia, a critical signaling event in microglial activation (92) that promotes pain signal amplification (Figure 1). Another potential pathway is through the receptors P2X4 and P2X7, which are upregulated in microglia after nerve injury and activated in response to injury by ATP released by primary sensory and dorsal horn neurons as well as dorsal horn astrocytes (94). P38, a mitogen-activated protein kinase (MAPK) has been implicated as a major participant in the activation of spinal microglia (FIG 1). Originally demonstrated in a neuropathic pain model using sciatic nerve ligation, Jin et al showed early p38 activation in spinal microglia (12-24 hrs after injury), with subsequent activation in DRG neurons (55). Inhibition of this activation with a p38-inhibitor also inhibited the development of pain hypersensitivity (93). P38 activation has also been reported in the rat model of chronic pancreatitis pain by Liu et al, suggesting that this pathway is important in sustained visceral pain (62). Once activated, these loops can function without further external stimulus. Thus, excitation of nociceptive spinal neurons leads to activation of spinal microglia via multiple parallel pathways that provide an efficient means to amplify inflammatory nociceptive signals.
How does microglial activation cause sustained pain? It has been well established that support cells of the nervous system participate in maintaining neuropathic pain pathways in somatic pain models (86, 93, 101). Activation of spinal microglia leads to the release of the soluble chemokine fractalkine (FKN) which is expressed in CNS sensory neurons as a transmembrane protein that can be cleaved to a soluble form (FIG 1). This cleavage was originally shown to occur after excitotoxic stimuli, suggesting that fractalkine cleavage represented an early event in the neurogenic inflammatory process (18). It is now known that membrane-bound FKN is cleaved by the cysteine protease cathepsin S (Cat S), which is secreted peripherally by macrophages (7) and centrally by activated microglia. Cat S cleaves FKN on dorsal horn neurons, releasing its soluble form, which then binds its own receptor CX3CR1 (21). This receptor is only expressed in microglia that are in the activated state (22) and binding further activates the p38 pathway in a positive feedback loop. Using the rat model of peripheral nerve ligation, Clark et al showed that using a Cat S inhibitor reduced pain behavior 7 and 14-days after sciatic nerve ligation in rats, whereas it did not prevent the initial development of pain. This suggests that Cat S is important in the maintenance of neuropathic pain, rather than the development of hyperalgesia (21). The release of Cat S, and subsequent binding and activation of the p38 pathway is dependent on microglial activation.
Recent evidence supports the importance of activated microglia in the development and maintenance of sustained visceral pain. In a rat model of TNBS-induced chronic pancreatitis, Liu et al found that the microglial activation inhibitor minocycline significantly decreased nociceptive behavior, and that withdrawal of minocycline caused a return to baseline. Also, pre-treatment with minocycline, prior to injection of TNBS, prevented chronic visceral hyperalgesia for as long as 3 weeks (62). We found similar results in the TNBS-induced chronic pancreatitis mouse treated with minocycline, with normalization of the expected heightened responses to Von Frey filament probing (unpublished results.) These data suggest that microglial activation may play an important role in sustained pancreatic pain.
Astrocytes
Astrocytes demonstrate activated morphology in neuropathic pain models, (42) and drugs used to treat pain in these experimental conditions attenuate activation (43, 101). Activation of astrocytes by mediators such as ATP, SP, prostaglandins, and glutamate, released by sensory nerves in response to injury, stimulates release of pro-inflammatory mediators. These mediators include cytokines such as IL-1B, IL-6 and TNF-a, as well as the molecules that activate them, including ATP and prostaglandins (101). Multiple intracellular signaling pathways have been implicated in the regulation of astrocyte activation, including p38, c-Jun-N-terminal kinase (JNK) and, perhaps most importantly, the extracellular signal-regulated kinase (ERK) pathway(54). Zhuang et al demonstrated increased expression of phosphorylated ERK in both microglia and dorsal horn astrocytes 10 days after spinal nerve ligation. In this study, intrathecal injection of an ERK inhibitor significantly reduced mechanical allodynia (109). In the rat TNBS chronic pancreatitis pain model, Feng et al reported an increase in glial fibrillary acidic protein (GFAP), an astrocyte marker that is upregulated in somatic models of neuropathic pain. This study also importantly demonstrated that attenuation of neuropathic pain in this model was possible using L-α-aminoadipate (LAA), a specific inhibitor of astrocyte activation, (38) suggesting a potential therapeutic target.
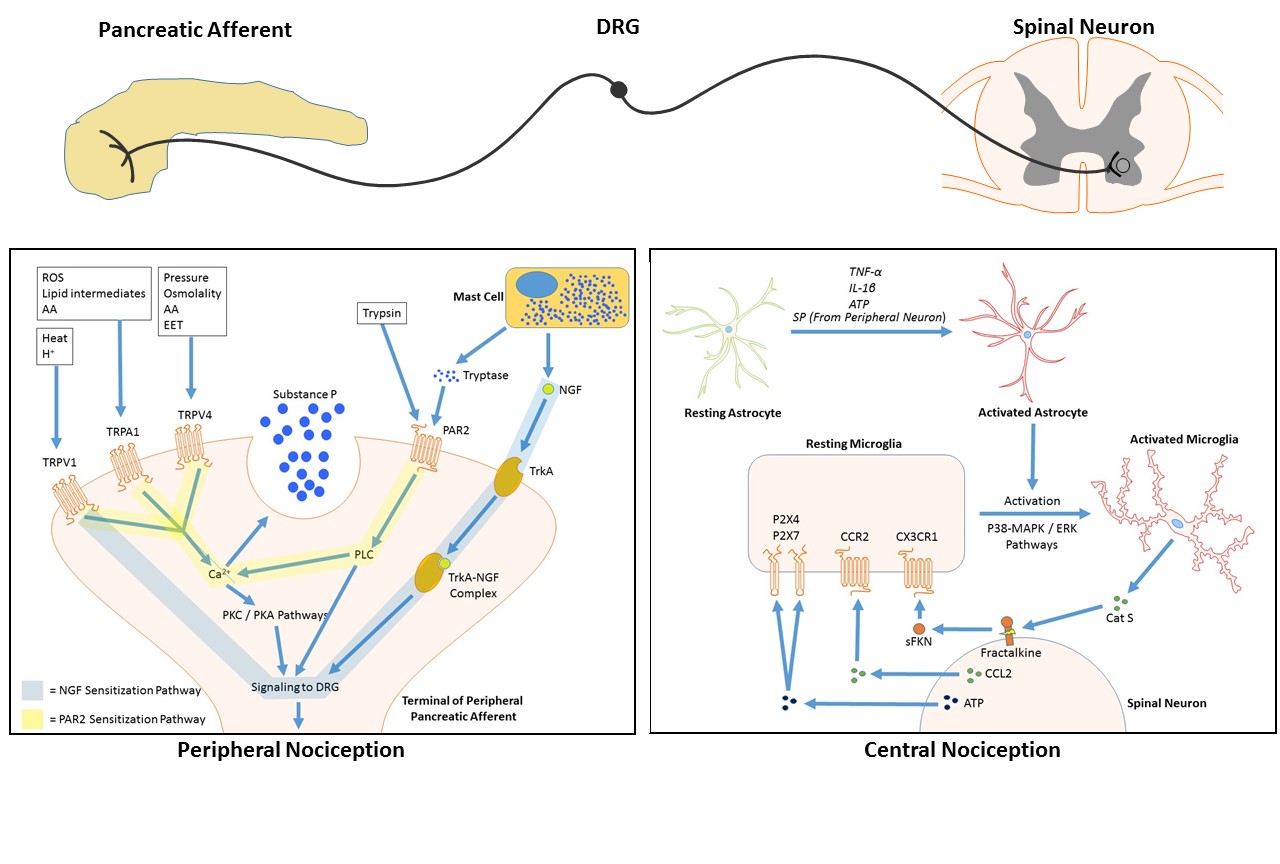
Figure 1: Pathways of pancreatic pain signal transmission in chronic pancreatitis with emphasis on mechanisms of sensitization.Peripherally, extracellular inflammatory agents including NGF, trypsin and tryptase sensitize and activate pancreatic afferent nociceptive neurons through integrative calcium signaling pathways. Centrally, sensitization is mediated through positive feedback loops among dorsal horn neurons and the activated neuronal supporting cells microglia and astrocytes, via Cat S-mediated cleavage and release of soluble FKN. Abbreviations: ROS, reactive oxygen species; AA, arachadonic acid metabolites; TRPV, transient receptor potential vanilloids; NGF, nerve growth factor; PAR2, protease activated receptor 2; PLC, phospholipase C; PKC, protein kinase C; PKA, protein kinase A; DRG, dorsal root ganglia; EET, epoxyeicosatrienoic acids; TrkA, trypo-myosin related kinase A; SP, substance P; FKN, fractalkine; sFKN, soluble fractalkine; Cat S, cathepsin S; CCR2, chemokine receptor 2; CCL2, chemokine ligand 2; MAPK, map kinase pathway; ERK, extracellular signal-related kinase pathway.
Reorganization
Observational studies in humans with chronic visceral pain have led to the notion that changes in inhibitory and amplification processes in the CNS contribute to reorganization of referred pain signal mapping. Mertz et al meticulously mapped pain patterns in patients with inflammatory bowel syndrome (IBS). In response to rectal distension, IBS patients had increased hypersensitivity in areas remote from the stimulus, as well as larger overall pain areas, as compared with healthy controls. These changes were associated with increased thalamic activation in the brain. They suggested that increased afferent signaling from the gut may lead to perceptual reorganization of pain signals (70). Interestingly, in contrast to these results, Dimcevski and colleagues reported that chronic pancreatitis patients had hypoalgesia in response to balloon distension of viscera surrounding the pancreas compared with healthy controls (31). It is worth noting that duodenal distension is less well established as a marker of referred visceral hyperalgesia than rectal distension, which could contribute to these results. In other studies by this group, electrical visceral pain stimulation in chronic pancreatitis patients was associated with reduced evoked potential latency in the brain, thereby suggesting that central modulation of pain pathways contributes to visceral hypersensitivity. Similar to prior findings in IBS patients, chronic pancreatitis patients also demonstrated an increase in the mean areas of referred pain following electrical visceral stimulation, suggesting reorganization of pain perception (30).
Opioid-Induced Hyperalgesia
Opioids are the mainstay of treatment for patients with severe chronic pancreatitis pain. As early as the 19th century, it was recognized that chronic opioid use leads not only to tolerance and physical and psychological dependence, but, ironically, also to increased sensitivity to painful stimuli, or opioid-induced hyperalgesia (OIH). Unlike opioid tolerance, OIH cannot be mitigated
by increased dosage regimens (58). Multiple studies in animal models have shown a reduction in mechanical and thermal thresholds to nociceptive stimuli with opioid treatment (16, 65, 96). Similarly, there have been multiple clinical studies in humans describing varying levels of hyperalgesic states among both patients and healthy controls treated with chronic opioids (20, 23, 48, 81). Much remains to be defined about the mechanisms of this hyperalgesia, but they are thought to be closely intertwined with the pathways of opioid tolerance. Pathways involving the N-methyl-D-aspartate receptor (NMDAR), spinal glutamate activity, protein kinase C activity and spinal dynorphin have all been implicated as vital to both tolerance and hyperalgesia (67). It is likely that this phenomenon contributes, in part, to the exasperation experienced by both patients and physicians at the progressive worsening of chronic pancreatitis pain experienced by some patients on escalating opioid dosages.
6. Conclusion
Research into the mechanisms of chronic pancreatitis pain has been accelerated by the recent availability of validated rat and mouse models, which provide interested investigators with a wider array of reproducible measures of experimental visceral pain. Emerging models illustrate the complexity, redundancy, interconnectedness, and plasticity, of chronic visceral pain pathways. Current treatments do not address underlying mechanisms of sensitization and amplification of both peripheral and central pain signals, which may, in part, explain the high level of medical and surgical treatment failure. In the periphery, integrative channels offer potentially high-leverage targets, as do positive feedback loops in the spinal cord, where selective inhibition could have profound beneficial results. The development of clinically-useful inhibitors of these potential targets is expected to improve both treatment effectiveness and quality of life for patients with debilitating chronic pancreatitis pain.
7. References
- Alessandri-Haber N, Dina OA, Joseph EK, Reichling D, Levine JD. A transient receptor potential vanilloid 4-dependent mechanism of hyperalgesia is engaged by concerted action of inflammatory mediators. J Neurosci 26(14):3864-3874, 2006. PMID: 16597741.
- Alessandri-Haber N, Joseph E, Dina OA, Liedtke W, Levine JD. TRPV4 mediates pain-related behavior induced by mild hypertonic stimuli in the presence of inflammatory mediator. Pain 118(1-2):70-79, 2005. PMID: 16213085.
- Ammann RW, Muellhaupt B. The natural history of pain in alcoholic chronic pancreatitis. Gastroenterology 116(5):1132-1140, 1999. PMID: 10220505.
- Baghdadi S, Abbas MH, Albouz F, Ammori BJ. Systematic review of the role of thoracoscopic splanchnicectomy in palliating the pain of patients with chronic pancreatitis. Surg Endosc 22(3):580-588, 2008. PMID: 18163168.
- Ballantyne JC, Mao J. Opioid Therapy for Chronic Pain. N Engl J Med 349(20):1943-1953, 2003. PMID: 14614170.
- Barbacid M. The Trk family of neurotrophin receptors. J Neurobiol 25(11):1386-1403, 1994. PMID: 7852993.
- Barclay J, Clark AK, Ganju P, Gentry C, Patel S, Wotherspoon G, et al. Role of the cysteine protease cathepsin S in neuropathic hyperalgesia. Pain 130(3):225-234, 2007. PMID: 17250968.
- Bautista DM, Pellegrino M, Tsunozaki M. TRPA1: A gatekeeper for inflammation. Annu Rev Physiol 75:181-200, 2013. PMID: 23020579.
- Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, et al. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 124(6):1269-1282, 2006. PMID: 16564016.
- Bornman PC, Marks IN, Girdwood AW, Berberat PO, Gulbinas A, Büchler MW. Pathogenesis of pain in chronic pancreatitis: ongoing enigma. World J Surg 27(11):1175-1182, 2003. PMID: 14574490.
- Bradley EL 3rd. Pancreatic duct pressure in chronic pancreatitis. Am J Surg 144(3):313-316, 1982. PMID: 7114368.
- Cahen DL, Gouma DJ, Nio Y, Rauws EA, Boermeester MA, Busch OR, et al. Endoscopic versus surgical drainage of the pancreatic duct in chronic pancreatitis. N Engl J Med 356(7):676-684, 2007. PMID: 17301298.
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288(5464):306-313, 2000. PMID: 10764638.
- Cattaruzza F, Amadesi S, Carlsson JF, Murphy JE, Lyo V, Kirkwood K, et al. Serine Proteases and Protease-activated Receptor 2 Mediate the Proinflammatory and Algesic Actions of Diverse Stimulants. Br J Pharmacol 171(16):3814-3826, 2014. PMID: 24749982.
- Cattaruzza F, Johnson C, Leggit A, Grady E, Schenk AK, Cevikbas F, et al. Transient receptor potential ankyrin 1 mediates chronic pancreatitis pain in mice. Am J Physiol Gastrointest Liver Physiol 304(11):G1002-G1012, 2013. PMID: 23558009.
- Célèrier E, Laulin J, Larcher A, Le Moal M, Simonnet G. Evidence for opiate-activated NMDA processes masking opiate analgesia in rats. Brain Res 847(1):18-25, 1999. PMID: 10564731.
- Ceppa E, Cattaruzza F, Lyo V, Amadesi S, Pelayo JC, Poole DP, et al. Transient receptor potential ion channels V4 and A1 contribute to pancreatitis pain in mice. American journal of physiology. Am J Physiol Gastrointest Liver Physiol 299(3):G556-G571, 2010. PMID: 20539005.
- Chapman GA, Moores K, Harrison D, Campbell CA, Stewart BR, Strijbos PJ. Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J Neurosci 20(15):RC87, 2000. PMID: 10899174.
- Chen Y, Yang C, Wang ZJ. Proteinase-activated receptor 2 sensitizes transient receptor potential vanilloid 1, transient receptor potential vanilloid 4, and transient receptor potential ankyrin 1 in paclitaxel-induced neuropathic pain. Neuroscience 193:440-451, 2011. PMID: 21763756.
- Chu LF, Clark DJ, Angst MS. Opioid tolerance and hyperalgesia in chronic pain patients after one month of oral morphine therapy: a preliminary prospective study. J Pain 7(1):43-48, 2006. PMID: 16414554.
- Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, et al. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci U S A 104(25):10655-10660, 2007. PMID: 17551020.
- Clark AK, Yip PK, Malcangio M. The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. J Neurosci 29(21):6945-6954, 2009. PMID: 19474321.
- Compton P, Charuvastra VC, Ling W. Pain intolerance in opioid-maintained former opiate addicts: effect of long-acting maintenance agent. Drug Alcohol Depend ;63(2):139-146, 2001. PMID: 11376918.
- Costamagna G, Bulajic M, Tringali A, Pandolfi M, Gabbrielli A, Spada C, et al. Multiple stenting of refractory pancreatic duct strictures in severe chronic pancreatitis: long-term results. Endoscopy 38(3):254-259, 2006. PMID: 16528652.
- D'Haese JG, Ceyhan GO, Demir IE, Tieftrunk E, Friess H. Treatment options in painful chronic pancreatitis: a systematic review. HPB (Oxford) 16(6):512-521, 2014. PMID: 24033614.
- Dai Y, Wang S, Tominaga M, Yamamoto S, Fukuoka T, Higashi T, et al. Sensitization of TRPA1 by PAR2 contributes to the sensation of inflammatory pain. J Clin Invest 117(7):1979-1987, 2007. PMID: 17571167.
- Demir IE, Tieftrunk E, Maak M, Friess H, Ceyhan GO. Pain mechanisms in chronic pancreatitis: of a master and his fire. Langenbecks Arch Surg 396(2):151-160, 2011. PMID: 21153480.
- Déry O, Corvera CU, Steinhoff M, Bunnett NW. Proteinase-activated receptors: novel mechanisms of signaling by serine proteases. Am J Physiol 274:C1429-C1452, 1998. PMID: 9696685.
- Di Sebastiano P, di Mola FF, Di Febbo C, Baccante G, Porreca E, Innocenti P, et al. Expression of interleukin 8 (IL-8) and substance P in human chronic pancreatitis. Gut 47(3):423-428, 2000. PMID: 10940282.
- Dimcevski G, Sami SA, Funch-Jensen P, Le Pera D, Valeriani M, Arendt-Nielsen L, et al. Pain in chronic pancreatitis: the role of reorganization in the central nervous system. Gastroenterology 132(4):1546-1556, 2007. PMID: 17408654.
- Dimcevski G, Schipper KP, Tage-Jensen U, Funch-Jensen P, Krarup AL, Toft E, et al. Hypoalgesia to experimental visceral and somatic stimulation in painful chronic pancreatitis. Eur J Gastroenterol Hepatol 18(7):755-764, 2006. PMID: 16772833.
- Díte P, Ruzicka M, Zboril V, Novotný I. A prospective, randomized trial comparing endoscopic and surgical therapy for chronic pancreatitis. Endoscopy 35(7):553-558, 2003. PMID: 12822088.
- Drewes AM, Krarup AL, Detlefsen S, Malmstrøm ML, Dimcevski G, Funch-Jensen P, et al. Pain in chronic pancreatitis: the role of neuropathic pain mechanisms. Gut 57(11):1616-1627, 2008. PMID: 18566105.
- Ebbehøj N, Borly L, Madsen P, Matzen P. Pancreatic tissue fluid pressure during drainage operations for chronic pancreatitis. Scand J Gastroenterol 25(10):1041-1045, 1990. PMID: 2263876.
- Ebbehøj N, Borly L, Bülow J, Henriksen JH, Heyeraas KJ, Rasmussen SG. Evaluation of pancreatic tissue fluid pressure measurements intraoperatively and by sonographically guided fine-needle puncture. Scand J Gastroenterol 25(11):1097-1102, 1990. PMID: 2274734.
- Ebbehoj N. Pancreatic tissue fluid pressure and pain in chronic pancreatitis. Dan Med Bull 39(2):128-133, 1992. PMID: 1611919.
- Ebbehøj N, Borly L, Madsen P, Matzen P. Comparison of regional pancreatic tissue fluid pressure and endoscopic retrograde pancreatographic morphology in chronic pancreatitis. Scand J Gastroenterol 25(7):756-760, 1990. PMID: 2396092.
- Feng QX, Wang W, Feng XY, Mei XP, Zhu C, Liu ZC, et al. Astrocytic activation in thoracic spinal cord contributes to persistent pain in rat model of chronic pancreatitis. Neuroscience 167(2):501-509, 2010. PMID: 20149842.
- Friess H, Zhu ZW, di Mola FF, Kulli C, Graber HU, Andren-Sandberg A, et al. Nerve growth factor and its high-affinity receptor in chronic pancreatitis. Ann Surg 230(5):615-624, 1999. PMID: 10561084.
- Gachago C, Draganov PV. Pain management in chronic pancreatitis. World J Gastroenterol 14(20):3137-3148, 2008. PMID: 18506917.
- Garcea G, Thomasset S, Berry DP, Tordoff S. Percutaneous splanchnic nerve radiofrequency ablation for chronic abdominal pain. ANZ J Surg 75(8):640-644, 2005. PMID: 16076323.
- Garrison CJ, Dougherty PM, Kajander KC, Carlton SM. Staining of glial fibrillary acidic protein (GFAP) in lumbar spinal cord increases following a sciatic nerve constriction injury. Brain Res 565(1):1-7, 1991. PMID: 1723019.
- Garrison CJ, Dougherty PM, Carlton SM. GFAP expression in lumbar spinal cord of naive and neuropathic rats treated with MK-801. Exp Neurol 129(2):237-243, 1994. PMID: 7957738.
- Georgiev G, Beltran del Rio M, Gruessner A, Tiwari M, Cercone R, Delbridge M, et al. Patient quality of life and pain improve after autologous islet transplantation (AIT) for treatment of chronic pancreatitis: 53 patient series at the University of Arizona. Pancreatology 15(1):40-45, 2015. PMID: 25455347.
- Grady EF, Yoshimi SK, Maa J, Valeroso D, Vartanian RK, Rahim S, et al. Substance P mediates inflammatory oedema in acute pancreatitis via activation of the neurokinin-1 receptor in rats and mice. Br J Pharmacol 130(3):505-512, 2000. PMID: 10821777.
- Grant AD, Cottrell GS, Amadesi S, Trevisani M, Nicoletti P, Materazzi S, et al. Protease-activated receptor 2 sensitizes the transient receptor potential vanilloid 4 ion channel to cause mechanical hyperalgesia in mice. J Physiol 578:715-733, 2007. PMID: 17124270.
- Gress F, Schmitt C, Sherman S, Ciaccia D, Ikenberry S, Lehman G. Endoscopic ultrasound-guided celiac plexus block for managing abdominal pain associated with chronic pancreatitis: a prospective single center experience. Am J Gastroenterol 96(2):409-416, 2001. PMID: 11232683.
- Hay JL, White JM, Bochner F, Somogyi AA, Semple TJ, Rounsefell B. Hyperalgesia in opioid-managed chronic pain and opioid-dependent patients. J Pain 10(3):316-322, 2009. PMID: 19101210.
- Hoogerwerf WA, Shenoy M, Winston JH, Xiao SY, He Z, Pasricha PJ. Trypsin mediates nociception via the proteinase-activated receptor 2: a potentially novel role in pancreatic pain. Gastroenterology 127(3):883-891, 2004. PMID: 15362043.
- Hoogerwerf WA, Zou L, Shenoy M, Sun D, Micci MA, Lee-Hellmich H, et al. The proteinase-activated receptor 2 is involved in nociception. J Neurosci 21(22):9036-9042, 2001. PMID: 11698614.
- Hoogerwerf WA, Gondesen K, Xiao SY, Winston JH, Willis WD, Pasricha PJ. The role of mast cells in the pathogenesis of pain in chronic pancreatitis. BMC Gastroenterol 5:8, 2005. PMID: 15745445.
- Hutter MM, Wick EC, Day AL, Maa J, Zerega EC, Richmond AC, et al. Transient receptor potential vanilloid (TRPV-1) promotes neurogenic inflammation in the pancreas via activation of the neurokinin-1 receptor (NK-1R). Pancreas 30(3):260-265, 2005. PMID: 15782105.
- Hwang SW, Cho H, Kwak J, Lee SY, Kang CJ, Jung J, et al. Direct activation of capsaicin receptors by products of lipoxygenases: endogenous capsaicin-like substances. Proc Natl Acad Sci U S A 97(11):6155-6160, 2000. PMID: 10823958.
- Ji RR, Kawasaki Y, Zhuang ZY, Wen YR, Decosterd I. Possible role of spinal astrocytes in maintaining chronic pain sensitization: review of current evidence with focus on bFGF/JNK pathway. Neuron Glia Biol 2(4):259-269, 2006. PMID: 17710215.
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci 23(10):4017-4022, 2003. PMID: 12764087.
- Kwan KY, Allchorne AJ, Vollrath MA, Christensen AP, Zhang DS, Woolf CJ, et al. TRPA1 contributes to cold, mechanical, and chemical nociception but is not essential for hair-cell transduction. Neuron 50(2):277-289, 2006. PMID: 16630838.
- Larsson LI. Innervation of the pancreas by substance P, enkephalin, vasoactive intestinal polypeptide and gastrin/CCK immunoractive nerves. J Histochem Cytochem 27(9):1283-1284, 1979. PMID: 479572.
- Lee M, Silverman SM, Hansen H, Patel VB, Manchikanti L. A comprehensive review of opioid-induced hyperalgesia. Pain Physician 14(2):145-161, 2011. PMID: 21412369.
- Lewin GR, Ritter AM, Mendell LM. Nerve growth factor-induced hyperalgesia in the neonatal and adult rat. J Neurosci 13(5):2136-2148, 1993. PMID: 8478693.
- Liddle RA. The role of Transient Receptor Potential Vanilloid 1 (TRPV1) channels in pancreatitis. Biochim Biophys Acta 1772(8):869-878, 2007. PMID: 17428642.
- Liedtke W, Friedman JM. Abnormal osmotic regulation in trpv4-/- mice. Proc Natl Acad Sci U S A 100(23):13698-13703, 2003. PMID: 14581612.
- Liu PY, Lu CL, Wang CC, Lee IH, Hsieh JC, Chen CC, et al. Spinal microglia initiate and maintain hyperalgesia in a rat model of chronic pancreatitis. Gastroenterology 142(1):165-173, 2012. PMID: 21963786.
- Lyo V, Cattaruzza F, Kim TN, Walker AW, Paulick M, Cox D, et al. Active cathepsins B, L, and S in murine and human pancreatitis. American journal of physiology. Am J Physiol Gastrointest Liver Physiol 303(8):G894-G903, 2012. PMID: 22899821.
- Mantyh PW, Mantyh CR, Gates T, Vigna SR, Maggio JE. Receptor binding sites for substance P and substance K in the canine gastrointestinal tract and their possible role in inflammatory bowel disease. Neuroscience (3):817-837, 1988. PMID: 2457186.
- Mao J, Sung B, Ji RR, Lim G. Chronic morphine induces downregulation of spinal glutamate transporters: implications in morphine tolerance and abnormal pain sensitivity. J Neurosci 22(18):8312-8323, 2002. PMID: 12223586.
- Mao J, Price DD, Mayer DJ. Mechanisms of hyperalgesia and morphine tolerance: a current view of their possible interactions. Pain 62(3):259-274, 1995. PMID: 8657426.
- Mao J. Opioid-induced abnormal pain sensitivity: implications in clinical opioid therapy. Pain 100(3):213-217, 2002. PMID: 12467992.
- McMahon SB, Armanini MP, Ling LH, Phillips HS. Expression and coexpression of Trk receptors in subpopulations of adult primary sensory neurons projecting to identified peripheral targets. Neuron 12(5):1161-1171, 1994. PMID: 7514427.
- McMahon SB, Bennett DL, Priestley JV, Shelton DL. The biological effects of endogenous nerve growth factor on adult sensory neurons revealed by a trkA-IgG fusion molecule. Nat Med 1(8):774-780, 1995.PMID: 7585179.
- Mertz H. Role of the brain and sensory pathways in gastrointestinal sensory disorders in humans. Gut 51 Suppl 1:i29-33, 2002. PMID: 12077061.
- Michaels AJ, Draganov PV. Endoscopic ultrasonography guided celiac plexus neurolysis and celiac plexus block in the management of pain due to pancreatic cancer and chronic pancreatitis. World J Gastroenterol 13(26):3575-3580, 2007. PMID: 17659707.
- Mullady DK, Yadav D, Amann ST, O'Connell MR, Barmada MM, Elta GH, et al. Type of pain, pain-associated complications, quality of life, disability and resource utilisation in chronic pancreatitis: a prospective cohort study. Gut 60(1):77-84, 2011. PMID: 21148579.
- Nilius B. TRP channels in disease. Biochim Biophys Acta 1772(8):805-812, 2007. PMID: 17368864.
- Obata K, Katsura H, Mizushima T, Yamanaka H, Kobayashi K, Dai Y, et al. TRPA1 induced in sensory neurons contributes to cold hyperalgesia after inflammation and nerve injury. J Clin Invest 115(9):2393-2401, 2005. PMID: 16110328.
- Olesen SS, Bouwense SA, Wilder-Smith OH, van Goor H, Drewes AM. Pregabalin reduces pain in patients with chronic pancreatitis in a randomized, controlled trial. Gastroenterology 141(2):536-543, 2011. PMID: 21683078.
- Pasricha PJ. Unraveling the mystery of pain in chronic pancreatitis. Nat Rev Gastroenterol Hepatol 9(3):140-151, 2012. PMID: 22269952.
- Petrus M, Peier AM, Bandell M, Hwang SW, Huynh T, Olney N, et al. A role of TRPA1 in mechanical hyperalgesia is revealed by pharmacological inhibition. Mol Pain 3:40, 2007. PMID: 18086313.
- Pezzilli R, Morselli Labate AM, Ceciliato R, Frulloni L, Cavestro GM, Comparato G, et al. Quality of life in patients with chronic pancreatitis. Dig Liver Dis 37(3):181-189, 2005. PMID: 15888283.
- Poole DP, Amadesi S, Veldhuis NA, Abogadie FC, Lieu T, Darby W, et al. Protease-activated receptor 2 (PAR2) protein and transient receptor potential vanilloid 4 (TRPV4) protein coupling is required for sustained inflammatory signaling. J Biol Chem 288(8):5790-5802, 2013. PMID: 23288842.
- Puig-Diví V, Molero X, Salas A, Guarner F, Guarner L, Malagelada JR. Induction of chronic pancreatic disease by trinitrobenzene sulfonic acid infusion into rat pancreatic ducts. Pancreas 13(4):417-424, 1996. PMID: 8899803.
- Reznikov I, Pud D, Eisenberg E. Oral opioid administration and hyperalgesia in patients with cancer or chronic nonmalignant pain. Br J Clin Pharmacol 60(3):311-318, 2005. PMID: 16120071.
- Riediger H1, Adam U, Fischer E, Keck T, Pfeffer F, Hopt UT, et al. Long-term outcome after resection for chronic pancreatitis in 224 patients. J Gastrointest Surg 11(8):949-959; discussion 959-960, 2007. PMID: 17534689.
- Savage SR. Long-term opioid therapy: assessment of consequences and risks. J Pain Symptom Manage 11(5):274-286, 1996. PMID: 8636626.
- Schwartz ES, La JH, Scheff NN, Davis BM, Albers KM, Gebhart GF. TRPV1 and TRPA1 antagonists prevent the transition of acute to chronic inflammation and pain in chronic pancreatitis. J Neurosci 33(13):5603-5611, 2013. PMID: 23536075.
- Shrikhande SV, Friess H, di Mola FF, Tempia-Caliera A, Conejo Garcia JR, Zhu Z, et al. NK-1 receptor gene expression is related to pain in chronic pancreatitis. Pain 91(3):209-217, 2011. PMID: 11275376.
- Smith HS. Activated microglia in nociception. Pain Physician 13(3):295-304, 2010. PMID: 20495595.
- Staruschenko A, Jeske NA, Akopian AN. Contribution of TRPV1-TRPA1 interaction to the single channel properties of the TRPA1 channel. J Biol Chem 285(20):15167-15177, 2010. PMID: 20231274.
- Steinhoff M, Vergnolle N, Young SH, Tognetto M, Amadesi S, Ennes HS, et al. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med 6(2):151-158, 2000. PMID: 10655102.
- Sutherland DE, Radosevich DM, Bellin MD, Hering BJ, Beilman GJ, Dunn TB, et al. Total pancreatectomy and islet autotransplantation for chronic pancreatitis. J Am Coll Surg 214(4):409-424; discussion 424-426, 2012. PMID: 22397977.
- Talukdar R, Murthy HV, Nageshwar Reddy D. Role of methionine containing antioxidant combination in the management of pain in chronic pancreatitis: A systematic review and meta-analysis. Pancreatology 15(2):136-144, 2015. PMID: 25648074.
- Tandan M1, Reddy DN, Talukdar R, Vinod K, Santosh D, Lakhtakia S, et al. Long-term clinical outcomes of extracorporeal shockwave lithotripsy in painful chronic calcific pancreatitis. Gastrointest Endosc 78(5):726-733, 2013. PMID: 23891416.
- Thacker MA, Clark AK, Bishop T, Grist J, Yip PK, Moon LD, et al. CCL2 is a key mediator of microglia activation in neuropathic pain states. Eur J Pain ;13(3):263-272, 2009. PMID: 18554968.
- Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in "small" glia. Trends Neurosci 28(2):101-107, 2005. PMID: 15667933.
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 424(6950):778-783, 2003. PMID: 12917686.
- Ueda J, Miyasaka Y, Ohtsuka T, Takahata S, Tanaka M. Short- and long-term results of the Frey procedure for chronic pancreatitis. J Hepatobiliary Pancreat Sci 22(3):211-216, 2015. PMID: 25339262.
- Vanderah TW, Ossipov MH, Lai J, Malan TP Jr, Porreca F. Mechanisms of opioid-induced pain and antinociceptive tolerance: descending facilitation and spinal dynorphin. Pain 92(1-2):5-9, 2001. PMID: 11323121.
- Vergnolle N, Bunnett NW, Sharkey KA, Brussee V, Compton SJ, Grady EF, et al. Proteinase-activated receptor-2 and hyperalgesia: A novel pain pathway. Nat Med 7(7):821-816, 2001. PMID: 11433347.
- Verhaegh BP, van Kleef M, Geurts JW, Puylaert M, van Zundert J, Kessels AG, et al. Percutaneous radiofrequency ablation of the splanchnic nerves in patients with chronic pancreatitis: results of single and repeated procedures in 11 patients. Pain Pract 13(8):621-626, 2013. PMID: 23301539.
- Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 64(6):1057-1068, 1991. PMID: 1672265.
- Walsh RM, Saavedra JR, Lentz G, Guerron AD, Scheman J, Stevens T, et al. Improved quality of life following total pancreatectomy and auto-islet transplantation for chronic pancreatitis. J Gastrointest Surg 16(8):1469-1477, 2012. PMID: 22673773.
- Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci 24(8):450-455, 2001. PMID: 11476884.
- Wehler M, Nichterlein R, Fischer B, Farnbacher M, Reulbach U, Hahn EG, et al. Factors associated with health-related quality of life in chronic pancreatitis. Am J Gastroenterol 99(1):138-146, 2004. PMID: 14687155.
- Wen YR, Tan PH, Cheng JK, Liu YC, Ji RR. Microglia: a promising target for treating neuropathic and postoperative pain, and morphine tolerance. J Formos Med Assoc 110(8):487-494, 2011. PMID: 21783017.
- Wick EC, Hoge SG, Grahn SW, Kim E, Divino LA, Grady EF, et al. Transient receptor potential vanilloid 1, calcitonin gene-related peptide, and substance P mediate nociception in acute pancreatitis. American journal of physiology. Am J Physiol Gastrointest Liver Physiol 290(5):G959-G969, 2006. PMID: 16399878.
- Winston JH, He ZJ, Shenoy M, Xiao SY, Pasricha PJ. Molecular and behavioral changes in nociception in a novel rat model of chronic pancreatitis for the study of pain. Pain 117(1-2):214-222, 2005. PMID: 16098667.
- Woolf, C.J., Safieh-Garabedian, B., Ma, Q.P., Crilly, P. & Winter, J. Nerve growth factor contributes to the generation of inflammatory sensory hypersensitivity. Neuroscience 62, 327-331 (1994).
- Zhu Y, Colak T, Shenoy M, Liu L, Pai R, Li C, et al. Nerve growth factor modulates TRPV1 expression and function and mediates pain in chronic pancreatitis. Gastroenterology 141(1):370-377, 2011. PMID: 21473865.
- Zhu Y, Mehta K, Li C, Xu GY, Liu L, Colak T, et al. Systemic administration of anti-NGF increases A-type potassium currents and decreases pancreatic nociceptor excitability in a rat model of chronic pancreatitis. American journal of physiology. Am J Physiol Gastrointest Liver Physiol 302(1):G176-G181, 2012. PMID: 22038828.
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain 114(1-2):149-159, 2005. PMID: 15733640.