
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2020.15
| Attachment | Size |
|---|---|
| 739.65 KB |
Abstract:
A key function of the exocrine pancreas is the production of digestive enzymes. The pancreatic acinar cell synthesizes, stores, and secretes the proenzymes and enzymes needed to digest dietary proteins, carbohydrates, and lipids. Meeting these functional requirements necessitates that the acinar cell has very high rates of protein synthesis and export. Nascent proteins undergo folding, select modifications, concentration, segregation from other classes of proteins, and vectorial movement before reaching their final destination in secretory (zymogen) granules. These are concentrated in the apical pole of the acinar cell. Eating stimulates neural and hormonal pathways that mediate acinar cell zymogen granule exocytosis into the pancreatic duct.
The exocrine pancreas has two major physiologic functions: it supplies the proenzymes and enzymes needed for digesting dietary lipids, carbohydrates, and proteins; and secretes a bicarbonate-rich fluid that neutralizes acidic gastric secretions and thus provides the correct pH for intestinal digestion by pancreatic enzymes. The acinar cell has been a model system for foundational studies of protein synthesis and export. After electron microscopy was developed, for example, cell biologists first visualized organelles and established their function by studying acinar cells (Figure 1). Here, we focus on acinar cell protein synthesis, trafficking, and processing in the pancreatic acinar cell necessary for its central role in producing digestive enzymes. We present results primarily obtained using rodent acinar cells, though the limited data from human acinar cells suggest the functions are likely the same as in rodents.
I. PROTEIN SYNTHESIS
A. The Ribosome
The ribosome is the central element of the protein synthetic machinery. The eukaryotic ribosome is composed of two subunits: a large 60S unit containing 28S, 5S, and 5.8S rRNA and approximately 49 proteins; and a small subunit at 40S which includes 18S rRNA and about 33 proteins. The two subunits form a groove wherein new protein synthesis directed by messenger RNA takes place, aided by associated protein complexes to initiate, elongate, and terminate protein synthesis. Secretory proteins, including pancreatic digestive enzymes, contain a distinct n-terminal signal sequence which contains a 15- to 50-amino acid peptide that includes a hydrophobic core. Also known as a leader sequence, the signal sequence allows nascent proteins to cross the endoplasmic reticulum (ER) membrane by traversing the translocon, a large multi-protein channel1. The signal sequence binds to the signal recognition particle complex (SRP) and brings the ribosome, attached nascent protein, and mRNA to the ER membrane SRP-receptor2. The SRP is then released and protein synthesis resumes with entry of the signal sequence into the translocon and its subsequent proteolytic cleavage from the nascent protein3. In addition to soluble export proteins, the translocon also mediates the insertion of transmembrane proteins into the ER membrane and has a role in protein degradation.
Figure 1. Electron micrograph of guinea pig pancreatic acinar cell showing compartment involved in nascent protein synthesis and storage. The vectorial features of this pathway from endoplasmic reticulum to secretory granule are evident. Nascent proteins are synthesized in about 5 min in the endoplasmic reticulum; then move by transport vesicles to the Golgi complex, where they exit at about 20 min in condensing vacuoles (a.k.a. immature secretory granules), and mature into secretory granules. (Image from reference 82).
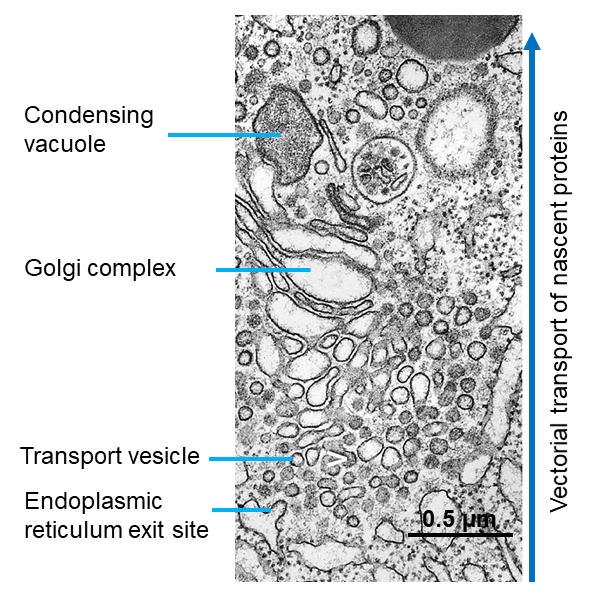
B. The Endoplasmic Reticulum (ER)
The synthesis of new secretory proteins occurs on and in the ER. This organelle has two morphologically distinct compartments: the rough ER (RER) and smooth ER (SER), which have specialized functions. The RER has ribosomes on its cytoplasmic surface which are easily seen by electron microscopy, whereas the SER lacks ribosomes (Figure 1). Between the RER and SER are regions termed transitional elements. Vesicular transport of nascent proteins from the ER occurs at sites in the transitional ER known as ER exit sites (ERES) (Figure 2). The intra-cisternal space is formed by RER, transitional elements, and SER; this is the site in which nascent proteins begin to fold and undergo export (see section 2 below). To accommodate high rates of secretory protein synthesis, a dense RER occupies much of the basal region of the acinar cell and extends apically. Regularly placed groupings of electron dense ribosomes [~30 nm] mark the sites of active protein synthesis on the cytosolic face of the ER membrane.
The acinar cell ER shows regional functional specialization that also includes cell signaling. For example, the initial acinar cell cytoplasmic calcium signal that mediates protein secretion depends on an ordered, spatial release from distinct ER stores. In response to neurohumoral stimulation, the first acinar cell signal arises at the apex of the cell from select ER domains and is mediated by the inositol triphosphate (IP3) receptor. Similarly, the propagation of the calcium wave throughout the acinar cell requires calcium release from ER ryanodine receptors which are distributed toward the base of the cell.
Distinct direct contacts between cell organelles have now been described in many tissues and include sites between the ER and mitochondria, and ER and plasma membrane. These are known to be involved in lipid transfer and calcium signaling4. In the acinar cell, apically-distributed IP3 receptors (ER) that release calcium into the cytosol are found to closely approximate regulators of calcium entry into the cell (Orai1, STIM, and others) that are activated when depletion of ER calcium is sensed5. These connections are thought to help coordinate both physiologic responses, such as protein secretion, and pathologic cell responses.
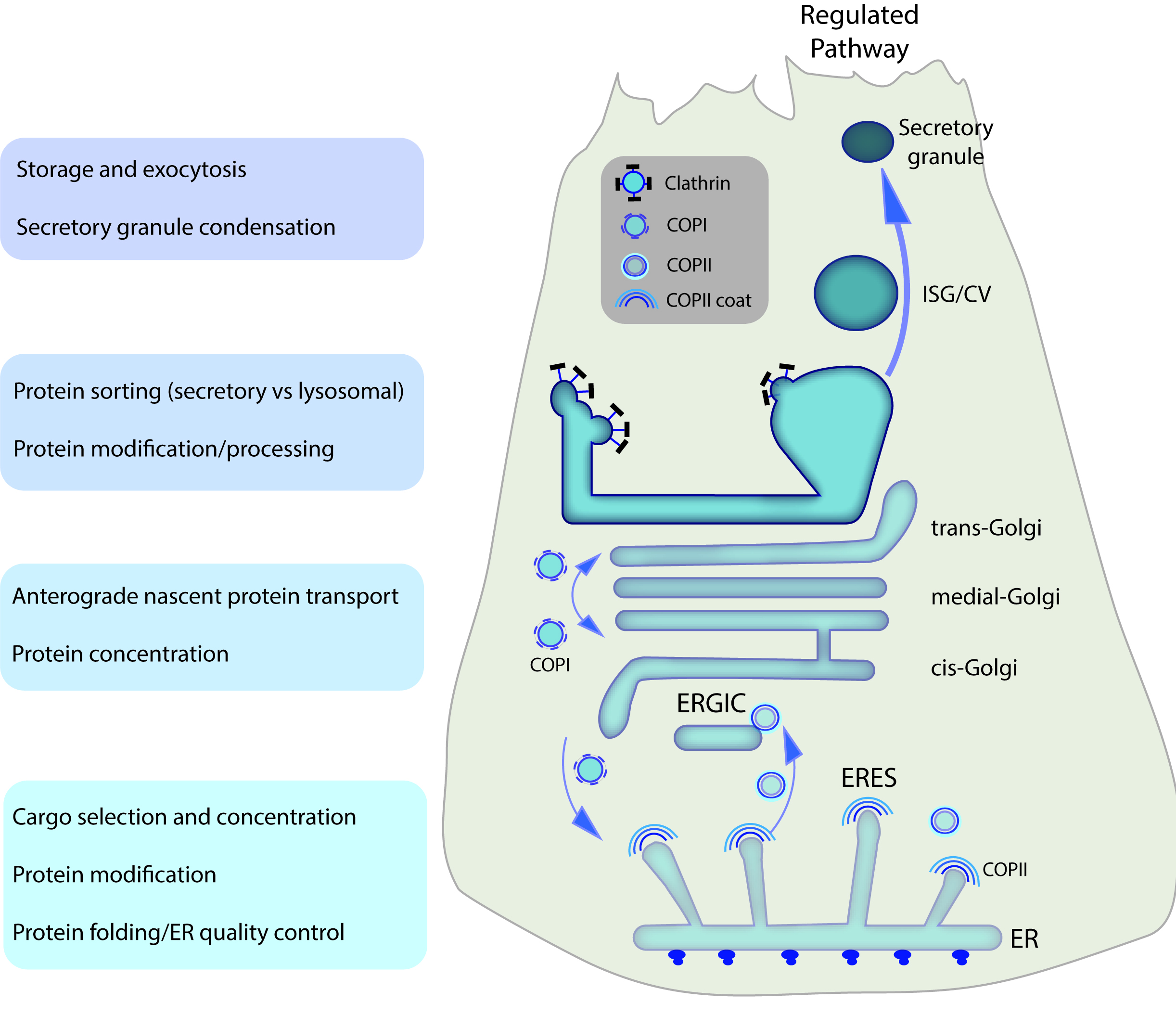
Figure 2. Secretory trafficking pathways in pancreatic acinar cells. Schematic depicting the classic secretory trafficking pathway in acini. Proteins are synthesized into the Endoplasmic Reticulum (ER) lumen, undergo folding and co-/post-translational modifications, and are selected and/or concentrated into COPII-coated vesicles at specialized ER exit sites (ERES). These vesicles interact with the ER-Golgi intermediate compartment (ERGIC), from which ER-resident proteins are sorted back to the ER via COPI-coated vesicles while secretory proteins are concentrated and trafficked through the Golgi stacks. Additional protein modifications occur in the Golgi. Secretory proteins are then packaged into immature secretory granules/condensing vacuoles (ISGs/CVs) through clathrin-mediated removal of membrane and cargo from the trans-Golgi; ISGs mature and condense by further removal of membrane/cargo. The resulting mature secretory granules are stored until signals for exocytosis are received by the cell.
II. MODIFICATIONS AND PROCESSING OF NASCENT PROTEINS
Nascent secretory proteins are subject to a variety of modifications; many of these occur within specific cellular organelles and are involved in forming the three-dimensional structure and covalent modifications necessary for proper protein function. Protein modifications often require the activity of accessory resident proteins that are concentrated in specific organelles.
A. Cleavage of the Signal Peptide
One of the first modifications of secretory proteins within the ER is the proteolytic removal of the signal peptide. The cleavage is mediated by a specific ER protease (signal peptidase) and occurs within the translocon after a large portion of the nascent protein has entered the ER cisterna3,6. Removal of the signal peptide traps the nascent protein in the secretory pathway unless the protein misfolds.
B. Protein Modifications and Folding
Secretory proteins undergo post-translational modifications within the ER that directs their folding into a tertiary structure that shields their hydrophobic residues in the interior of the molecule and forms their functional domains. Several major protein modifications, some of which are covalent, occur within the ER – including disulfide bond formation, glycosylation, and acetylation. Accessory ER resident proteins also direct the folding of nascent proteins.
Disulfide bonds
Disulfide bonds form by oxidative linkage of sulfhydryl groups between two Cys residues. These bonds are necessary to form higher order structures both within and between polypeptides and can occur either co- or post-translationally. The ER redox environment is uniquely suited to promote disulfide bond formation; indeed, disulfide bonds are highly abundant among secretory proteins and rarely found in cytosolic proteins. It follows that changes in ER redox status affect protein folding and stability. For example, proteomic assessment of acinar ER proteins following ethanol feeding shows a shift towards increased oxidation, which suggests the likelihood of aberrant disulfide bonds – leading to protein misfolding and ER stress7,8. That said, disulfide bonds can occur spontaneously under normal conditions, and so ER-resident protein disulfide isomerases (PDIs) act to rearrange disulfide bonds and ensure their formation between the correct Cys residues. As such, PDIs are also considered chaperones (see below). One PDI family member specifically expressed in pancreatic acinar cells, termed PDIp, interacts with several digestive enzymes and proteolytic zymogens, and prevents their aggregation9–11.
Glycosylation
Glycosylation describes the addition of carbohydrate chains called glycans to nascent proteins. Protein glycosylation in the ER is mediated by resident glycosyltransferases and glycosidases which generate a milieu of glycan moieties that can influence protein folding and function (reviewed in 12). One such modification is the addition of N-acetylglucosamine (GlcNAc) to Asn residues, termed N-glycosylation. N-glycosylation of newly translated peptides is regulated by the oligosaccharyltransferase (OST) complex by recognition of an Asn-X-(Thr/Ser) consensus sequence and occurs co-translationally via interactions between the OST and the translocon. Though N-glycosylation is understood to be critical for nascent protein folding, little is known about this particular modification in acinar cells. Studies in the early 1990s demonstrated the presence of N-glycosylation on human pancreatic elastase 1 (Ela1)13. It was later shown that N-glycosylation of bile salt-dependent lipase (BSDL) was essential for its expression, association with molecular chaperones, and secretion14,15. More work has been done investigating the significance of O-glycosylation of acinar cell proteins, which occurs in the Golgi, including lysosome-associated membrane proteins and some digestive enzymes (see section IV).
Acetylation
Studies over the last decade have identified a critical role for Nε-lysine acetylation and ER proteostasis; that is, the dynamic regulation of the biogenesis, folding, trafficking, and degradation of proteins within the ER. Post-translational acetylation of proteins in the ER is regulated by the ER acetyl-CoA transporter, AT-1 (SLC33A1), and ER-resident acetyltransferases ATase1 and ATase2 (NAT8B and NAT8, respectively), which utilize the luminal acetyl-CoA as acetylation substrate. In contrast to N-glycosylation, protein acetylation occurs only after successful protein folding and does not appear to involve a consensus sequence; though only outward-facing lysines are acetylated, suggesting a dependence on protein tertiary structure formation. Interestingly, the acetyltransferases interact with the OST complex to acetylate the targeted residues, suggesting a relationship between N-glycosylation and acetylation. The current view is that this acetylation identifies properly folded (i.e. glycosylated) proteins and promotes their entry into the secretory pathway, whereas improperly folded proteins cannot be acetylated appropriately and are thus subject to degradation. It has been recently shown that AT-1 expression increases at the onset of pancreatitis but falls as the disease progresses16. Furthermore, studies involving mice expressing mutant AT-1S113R/+, a human mutation associated with hereditary spastic paraplegias17, or pancreatic acinar-specific AT-1 deletion, show elevated ER stress, inflammation, and fibrosis consistent with a chronic pancreatitis-like phenotype, which unexpectedly includes enhanced trypsin activation16. Prior analyses of the so-called “ER acetylome” in neuronal cells identified acetylated proteins that are of interest in pancreatic acinar physiology, including BiP (GRP78), LAMP2, and cathepsin D18. Given the recently established role of AT-1 in pancreatic acinar homeostasis, investigating the acetylation state of acinar cell proteins will provide further insight into the functional significance of this modification on pancreatic outcomes.
Molecular chaperones
Although very small peptides can spontaneously fold unassisted, the folding of larger and more complex proteins in the ER is hindered by the available space and necessity of precise luminal conditions. ER-resident chaperones assist newly translated polypeptides by recognizing and interacting with key protein regions and shielding them from outside interference while the necessary modifications are made. Two main chaperone systems regulate this process in the ER: heat shock chaperones (e.g. BiP, aka GRP78, and GRP94), which interact with exposed hydrophobic regions; and lectins (e.g. calreticulin/calnexin), which interact with N-linked glycans. Evidence shows that these chaperones often cycle between association, dissociation, and reassociation with their targets until folding is complete. It follows that molecular chaperones are critical for pancreatic acinar cell function. In mice, GRP78+/- pancreas exhibits alterations in ER morphology, reduction in the expression of calreticulin and calnexin, and experience increased disease severity in response to caerulein-induced pancreatitis19. As previously mentioned, PDIs are also considered chaperones as they support protein folding by facilitating disulfide bond formation.
C. The Unfolded Protein Response (UPR)
To sustain high rates of ER protein synthesis and secretory trafficking efficiency, pancreatic acini rely on a robust ER stress response system to manage the accumulation of proteins in the ER lumen. Known as the unfolded protein response (UPR), this system is regulated by three ER transmembrane proteins that “sense” the load of unfolded proteins in the ER lumen: inositol requiring enzyme 1 (IRE1), activating transcription factor 6 (ATF6), and protein kinase R-like ER kinase (PERK). These sensors mediate signal transduction pathways that initiate cellular response mechanisms to alleviate the burden of ER stress (reviewed in 20,21).
The adaptive UPR
During the initial stages of ER stress, or when ER stress is mild, IRE1 dimerizes and auto-transphosphorylates, activating its RNase activity which processes the transcription factor X-box binding protein 1 (XBP1) into its spliced product (XBP1s)22,23. Effects of IRE1 on other types of mRNA have also been reported. Additionally, ATF6 translocates to the Golgi using the COPII complex coated vesicles and is cleaved by site 1 and 2 proteases to generate its active cytosolic form cATF624,25. Both XBP1s and cATF6 regulate the expression of genes encoding ER chaperones, ER biogenesis, and ER-associated degradation (ERAD); furthermore, cATF6 controls XBP1 expression22,23,26–29. These actions serve to increase the ER folding capacity and secretory output to clear accumulated proteins and are collectively referred to as the adaptive UPR.
The role of XBP1s in this protective mechanism has been extensively studied in the context of exocrine pancreas physiology and pancreatitis. XBP1s works in concert with the transcription factor Mist1 to drive terminal differentiation of pancreatic acini and other secretory cell types; XBP1 null mice are not viable28,30–35. XBP1 heterozygotes (XBP1+/-) exhibit increased pancreatic pathology, including oxidative stress that affects disulfide bond formation, in response to ethanol-mediated ER stress, which establishes a protective role for XBP1s during pancreatitis7,35. Moreover, XBP1s regulates the expression of AT-1 which is part of the ER protein acetylation machinery36. Although less is known about the role of ATF6 in acinar cells, a recent publication suggests a putative role for ATF6 and the apoptotic protein p53 in the development of chronic pancreatitis37.
The pathological UPR
In the event the adaptive UPR cannot ameliorate existing ER stress, subsequent pathways with more profound consequences are activated. Similar to IRE1, PERK autophosphorylates in response to ER stress and activates its kinase activity, which in turn phosphorylates the translation initiation factor 2α (eIF2α), inhibiting global cap-dependent translation. This provides the ER a reprieve from translational protein input. Should these actions fail to alleviate ER stress, PERK activates the apoptotic protein CCAAT/enhancer binding protein homologous protein (CHOP) by increasing expression of activating transcription factor 4 (ATF4). This duality in PERK function is illustrated by studies showing the necessity of both PERK and ATF4 in normal pancreatic development and function, whereas loss of CHOP results in normal exocrine function and protects against the acceleration of pancreatitis38–40. Sustained IRE1 signaling may also play a role in the pathological UPR by activating jun N-terminal kinase (JNK), which contributes to inflammation and apoptosis, though this pathway has not been fully studied in pancreatic acinar cells.
Regulated IRE1-dependent decay (RIDD)
Under conditions of sustained ER stress, the endoribonuclease activity of IRE1 may become broader, affecting the translation of mRNAs other than XBP1. Reports indicate a potential role for RIDD in regulating insulin mRNA in pancreatic β cells under ER stress due to hyperglycemia41,42. The physiological role of RIDD in exocrine pancreatic function and response to stress has not yet been explored.
D. Removal and Degradation of Accumulated Proteins
ER-associated degradation (ERAD) is a cellular pathway that removes terminally misfolded proteins from the ER lumen or membrane and regulates their degradation. ERAD may occur by proteasomal (ERAD-I) or autophagic (ERAD-II) mechanisms. It is believed that ERAD-I mediates the disposal of monomeric proteins. The best characterized branch of ERAD-I is regulated by the adaptor protein suppressor/enhancer of Lin-12-like (Sel1L) and the dislocon channel HMG-coA reductase degradation protein (Hrd1) complex, which retrotranslocates misfolded ER proteins to the cytosol where they are subsequently ubiquitinylated and targeted to the proteasome for degradation43. Interestingly, inducible Sel1L knockout mice exhibit classic exocrine insufficiency as well as persistent ER stress/UPR and, curiously, significantly smaller zymogen granules44. This study suggests a key role for Sel1L and ERAD-I in pancreatic acinar ER homeostasis.
ER homeostatic mechanisms utilizing autophagic/lysosomal pathways will be collectively referred to here as ERAD-II, including ER-phagy, reticulophagy, and/or ER-quality control (ERQC) autophagy; whether these classifications represent similar or distinct processes is the subject of debate which will not be dissected here. In contrast to ERAD-I, ERAD-II is proposed to degrade large protein aggregates and ER membrane rather than individual proteins (reviewed in 45). In mammals, autophagy-mediated ER degradation utilizes ER membrane associated receptor molecules. To date, six receptors have been identified: RTN3L, FAM134B, SEC62, CCPG1, ATL3, and TEX264. Although the various stimuli and protein targets for these receptors have not been completely characterized, they all interact with the autophagy protein LC3 through LC3 interacting regions (LIRs) which mediates the selective removal of ER through the autophagy pathway. Cell-cycle progression gene 1 (CCPG1) was first identified as an ER-phagy cargo receptor in pancreatic acini that is induced by UPR signaling, and loss of CCPG1 leads to ER disordering and loss of polarity in acini46–48.
Initiation of ERAD-II may be regulated by acetylation. The autophagy regulatory protein ATG9 is the only membrane-associated autophagy protein that resides in the ER. Studies of AT-1 function identified acetylation sites on ATG9 on the luminal side which restricts its activity, and loss of AT-1 function spurs increased autophagy36,49. These findings suggest that ATG9 may act as an ER lumen acetylation sensor wherein loss of acetyl-CoA availability, corresponding to accumulation of proteins, induces reticulophagy.
The extent to which ERAD-I and ERAD-II regulate the degradation of pancreatic acinar cell proteins under normal and disease states is unclear. Similarly, whether defects in either ERAD mechanism contribute to aberrant intracellular trypsin activation and/or pancreatitis pathology remains to be investigated.
III. EARLY ACINAR CELL SECRETORY PROTEIN TRAFFICKING
Pancreatic acinar cell secretory proteins are translated into the highly expanded rough ER (Figure 1, 2). Following folding and post-translational modifications discussed above, soluble and membrane-associated secretory proteins adjacent to specialized ERES are packaged into coat protein complex II (COPII)-coated vesicles and directed to the ER-Golgi intermediate compartment (ERGIC). In the ERGIC, resident ER proteins are sorted back to the ER in COPI-coated vesicles, while secretory proteins are concentrated and delivered to the Golgi cisternae (cis-, medial- and trans-Golgi), sometimes termed the Golgi stacks and/or Golgi ribbons (reviewed in 50–52). Proteins may undergo further posttranslational modifications during their sequential movement through the Golgi. Ultimately, secretory proteins are concentrated into condensing vacuoles within the trans-Golgi compartment where they bud off as precursors of secretory or zymogen granules (ZGs). It should be noted that condensing vacuoles (CV) and immature secretory granules (ISGs) are often used interchangeably in acinar cell literature (Figure 2, 3).
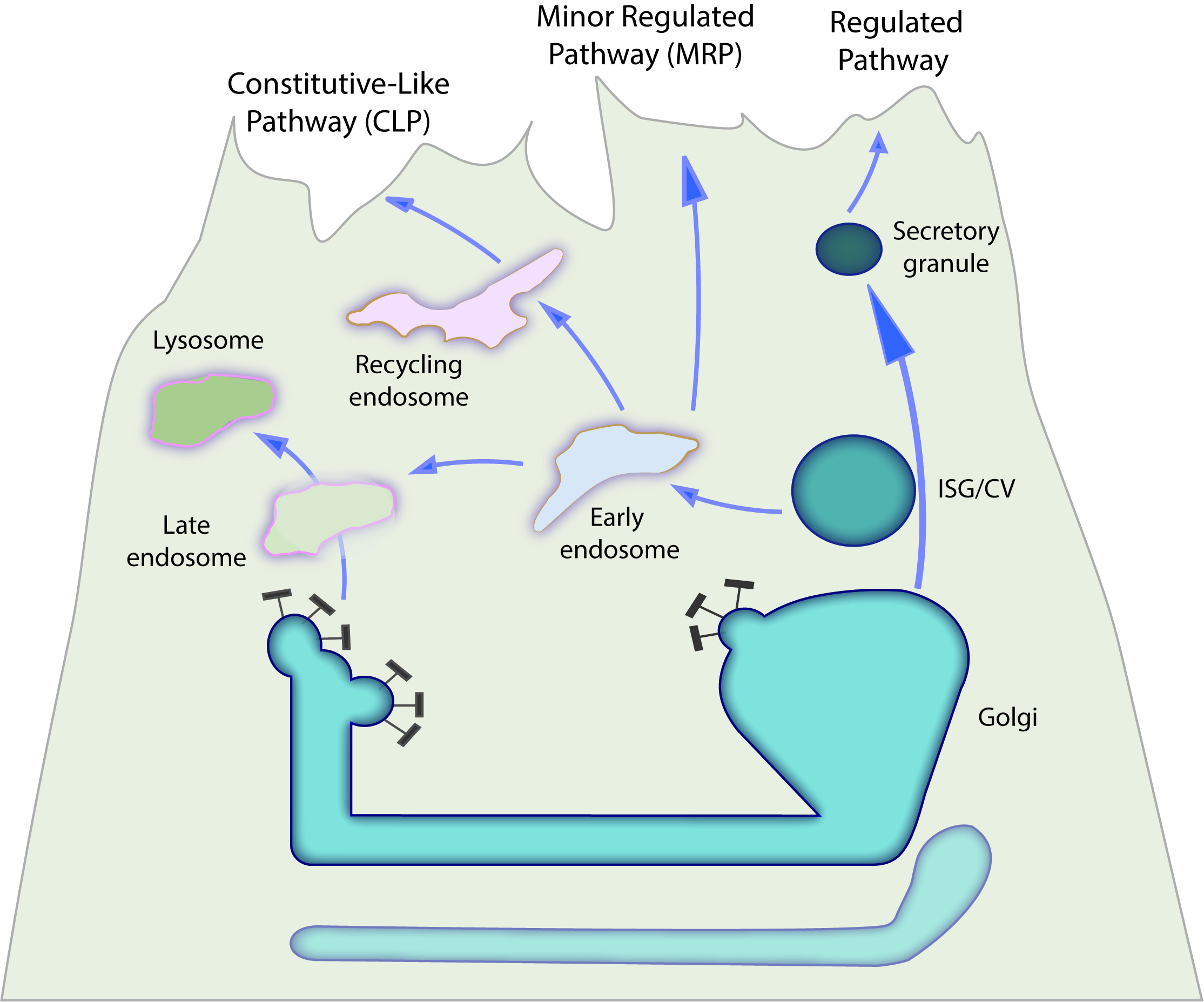
Figure 3. Additional secretory pathways identified in acinar cells. The classic regulated secretory pathway is represented by the formation of mature secretory granules from ISGs/CVs. Parallel secretory pathways are formed by the trafficking of membrane and secretory proteins through endosomal compartment intermediates prior to secretion, either directly from the early endosome in the minor-regulated pathway (MRP), or through the recycling endosome, as in the constitutive-like pathway (CLP).
A. ER to Golgi Trafficking.
Anterograde transport of ER membrane and protein to the ERGIC begins with the formation of COPII vesicles at specialized ERES subdomains51,53 . Formation of the COPII coat is directed by the cytosolic proteins Sar1, Sec23, Sec24, Sec13, and Sec3152,54. In brief, the GTPase Sar1 is GTP-loaded and activated by the guanine exchange factor Sec12 near the ERES, prompting the insertion of Sar1 into the ER membrane using an amphipathic helix. Sar1 membrane insertion facilitates the membrane deformation ultimately required for vesicle formation and budding. Membrane-associated Sar1 also binds to the heterodimer Sec23-Sec24. Sec 24 interacts with transmembrane ER-associated receptors to facilitate soluble cargo loading. Sequential formation of these complexes at the ERES promotes the addition of an outer layer composed of a heterotetramer of two Sec13 and two Sec31 subunits which self-assemble into cage-like structures morphologically similar, but biophysically distinct to, clathrin coats55. In addition to the five core COPII regulatory proteins, a growing number of proteins that transiently interact during COPII coat formation and trafficking have been identified but will not be detailed here52,54. Vesicle scission occurs through an unclear process, and the COPII vesicles – typically 60-80 nm in diameter – are directed to the ERGIC where they tether and undergo fusion via soluble n-ethylmaleimide sensitive receptor (SNARE) protein interactions56.
Although the acinar cell is known for its massive protein secretory capacity and many studies have been directed at understanding the mechanisms of COPII vesicle formation in other cells, few have addressed this pathway in pancreatic acinar cells. In 1999, Martínez-Menárguez et al. used immuno-electron microscopy on pancreas thin sections to demonstrate that the membrane-associated SNARE protein rBet1 is concentrated in COPII coated vesicles; however, the soluble secretory proteins amylase and chymotrypsin were not, suggesting that soluble cargo loading is not receptor-mediated but occurs by nonselective transport57. A later study in yeast followed the loading of soluble secretory proteins and COPII membrane proteins demonstrating receptor-mediated concentrative sorting of soluble proteins into COPII vesicles58. Whether additional acinar secretory proteins other than amylase and chymotrypsin may be selectively loaded in COPII vesicles remains unknown. A more recent study demonstrated that pancreas-specific deficiency of the Sec23B isoform, but not Sec23A, results in embryonic lethality due to acinar cell degeneration but, interestingly, did not disrupt the morphology of islets59,60. Clearly, further investigation of COPII formation, loading, and trafficking in normal acinar cells and the importance of this pathway during pathologic ER stress is warranted.
B. ER Golgi Intermediate Compartment (ERGIC)
The ERGIC, also known as the vesicular tubular cluster due to its morphology, acts as sorting compartment between the ER and Golgi apparatus (Figure 1, 2). The most reliable marker of this compartment is ERGIC-53, a mannose-specific membrane lectin that functions as a cargo receptor for the transport of glycoproteins from the ER to the ERGIC61. The ERGIC is formed and maintained by the continuous fusion of COPII vesicles arriving from the ER. Resident ER proteins are retrieved from the ERGIC back to the ER by their concentration and sorting into COPI-coated vesicles. In contrast to COPII coat formation, COPI coat proteins are composed of seven COP adapters that are assembled in response to activation of an ADP ribosylation factor (Arf1, Arf2 or Arf3) by GTP loading with guanine exchange factors. Activation triggers Arf myristoylation and membrane tethering identical to Arf function in clathrin coat formation (reviewed in 62). Cargo selection by COPI is directed by sorting signals present on cytoplasmic domains including di-lysine, KKxx, and KxKxx motifs present on many ER proteins. Likewise, the KDEL receptor, which functions to both retain ER resident proteins and recycle them from the ERGIC and cis-Golgi, interacts with KDEL-containing proteins, including ER chaperones, thereby concentrating them in COPI vesicles56.
In acinar cells, Martínez-Menárguez et al. showed that although amylase and chymotrypsin were not selectively sorted during COPII vesicle formation in the ER, these enzymes were concentrated in the ERGIC by their apparent exclusion from COPI vesicles recycling back to the ER57. This and a later study support the concept that the ERGIC functions as an initial concentrating compartment for acinar secretory proteins that are non-selectively transported from ER56,63. Interestingly, although COPI buds are most numerous in the ERGIC, they are also found in cisternae at all levels of the Golgi stacks as well as a minor presence on ISGs, indicating that either COPI recycling takes place at all levels of the early secretory pathway or that COPI vesicles have additional functions in the Golgi (see below)57. Of note, an earlier study by Sesso et al. provided a three-dimensional reconstruction of the rough ER-Golgi interface in serial thin sections of rat pancreas. This was depicted as a series budding small vesicles that fused to form tubulo-vesicular elements that appeared to be interposed between the ER and cis-Golgi, although the nature of these tubulo-vesicles is uncertain, it seems plausible they represented what is now termed the ERGIC64.
C. Golgi Apparatus (Complex)
A number of theories have been postulated regarding Golgi formation, structural organization, and mechanisms of protein and membrane trafficking between the Golgi compartments65–68. Golgi architecture in mammalian cells consists of a morphologically heterogeneous set of membrane-limited compartments with a characteristic stack-like appearance (Figure 1). Membrane and proteins generally flow or mature in an anterograde direction from cis- to trans-cisternae. The cis-cisternae are oriented towards the ER and are associated with small vesicle formation, whereas the trans-Golgi is oriented towards the apical plasma membrane and is associated with production of secretory granules. These regions are most commonly described as zones of the cis-, medial- and trans-Golgi, with movement between compartments by vesicle fission and fusion and so-called noncompact zones that are interconnected by lateral tubules between cisternae69,70. These interconnected regions appear as higher order structures by light microscopy giving rise to the term Golgi ribbon. There are numerous small, coated vesicles oriented both close to and budding off from the rims of the Golgi cisternae, the majority identified as COPI-coated by immuno-electron microscopy. COPI coats are most abundant in the ERGIC and cis-Golgi which decrease in number moving toward the trans-Golgi69. Recent studies have described these as COP1b vesicles that mediate anterograde trafficking within the Golgi cisternae whereas the COPIa vesicles mediate retrograde recycling of ER proteins from the ERGIC and cis-Golgi, although the molecular determinants dictating COPIa versus COPIb coats are uncertain56,67.
The most distal trans-Golgi cisternae give rise to and are continuous with the trans-Golgi network (TGN), a series of branching tubules with many budding profiles and forming vesicles. The TGN is biochemically distinct from the rest of the Golgi complex, having a higher concentration of certain proteins like TGN-38 compared to the cis- and medial-Golgi. The TGN is highly dynamic in accordance with the rates of protein synthesis and secretory protein entry into the compartment. Multiple organelles have been shown to arise from the TGN including early, late, and recycling endosomes, as well as vesicles specifically destined for apical and basolateral plasma membrane or back to the ER69. Unique to the TGN is large number of clathrin coats and associated clathrin-coated vesicles (Figure 3). Clathrin coat formation at Golgi membrane is mediated by assembly of clathrin adaptor protein complexes AP1, AP3, and AP4; these multisubunit complexes associate with cytoplasmic domains of specific Golgi transmembrane proteins to nucleate clathrin coat assembly71,72. In addition to heteromeric AP complexes, additional monomeric clathrin adaptors are present at the trans-Golgi including Golgi-localized γ-ear-containing Arf-binding proteins (GGAs) and enthoprotin/epsinR5673.
IV. PROTEIN MODIFICATION IN THE GOLGI COMPLEX
The Golgi is a major site of protein modification including terminal glycosylation, proteolytic processing, and sulfation, The Golgi fulfils its multiple functions using several classes of processing enzymes. These are primarily membrane proteins and may be regulated by their distinct localizations within the Golgi complex. For example, galactosyltransferase is restricted to the two or three trans-most Golgi cisternae74.
A. O-Glycosylation
Complex O-linked oligosaccharides are attached by glycotransferases within the Golgi (as opposed to N-linked glycosylation in the ER, see section II above). Lysosomal enzymes, including lysosome-associated membrane proteins 1 and 2 (LAMP1,2), are glycoproteins that are modified in this manner, and are essential for the function and integrity of lysosomes75. Furthermore, a study perturbing O-glycosylation in pancreas showed that a number of pancreatic digestive enzymes (e.g. bile salt-activated lipase, pancreatic triacylglycerol lipase, pancreatic alpha-amylase) are O-glycosylated, and that loss of O-glycosylation results in exocrine and endocrine insufficiency76.
B. Mannose-6-P Modification
In the cis-Golgi, mannose-6-phosphate (M6P) residues are added to proteins to direct sorting into the endolysosomal pathway74. These residues interact with the M6P receptor (MPR) which are localized to the Golgi region of polarized cells, coated vesicles, endosomes, and lysosomes as identified by Bron & Farquhar in 198477. Although an early study found MPRs to be restricted mainly to the cis-Golgi stacks, later work in acinar cells demonstrated AP1-mediated clathrin-coated vesicle retrieval of MPRs from immature secretory granules (see below) 77,78. Two distinct MPRs, a 46kDa MRP46 (cation-dependent MPR) and a 300 kDa MPR300 (cation-independent MRP) have been identified. Although the majority of soluble acid hydrolases are modified with M6P residues allowing their recognition by MPRs, other soluble enzymes and non-enzymatic proteins are transported to lysosomes in a M6P-independent manner mediated by alternative receptors such as lysosomal integral membrane protein (LIMP-2) or sortilin79. Sorting of cargo receptors and lysosomal transmembrane proteins requires sorting signals present in cytosolic domains which interact with components of clathrin coats or an adaptor protein complex. Additionally, phosphorylation and lipid modifications can further regulate signal recognition and trafficking79.
V. TRAFFICKING FROM THE GOLGI COMPLEX OUTWARD
A. Condensing Vacuoles and Formation of Zymogen Granules
The trans-Golgi is the site of condensing vacuole (or immature secretory granule, ISG) formation in pancreatic acinar cells. Early radio-labeling of acinar secretory proteins in vivo combined with electron microscopy radiographic analysis suggested the condensing vacuoles received secretory proteins directly transported on vesicular carriers from the so-called “ER transitional zone” which likely represented the ERES or ERGIC (Figure 1)80. Later higher resolution studies by Jameson and Palade in pancreatic slices from guinea pig stimulated with secretagogue revealed the presence of label in the Golgi stacks81; although in his 1974 Nobel acceptance speech Palade still depicted the ER transitional compartment as a direct route to the condensing vacuoles82. Though many studies have examined condensing vacuole formation, a comprehensive understanding of how secretory cargo are concentrated into condensing vacuoles, how enlarged vacuoles form, and the molecular mechanisms of condensing vacuole fission from the TGN remains uncertain but is likely to be driven at least in part by the acidification of the compartment.
The details of secretory granule formation in specialized secretory cells has received considerable attention over the last 30 years. Evidence derived from in vitro fusion assays in subcellular fractions of neuroendocrine cells supports that newly formed ISGs undergo homotypic fusion in a process mediated by syntaxin 6 and synaptotagmin IV83,84. Most studies in acinar cells posit that ISGs formed in the TGN are the direct precursors of mature secretory granules. However, Hammel et al.85 using a detailed morphometric analysis, proposed that small Golgi-derived vacuoles fuse to form ISGs85. They further propose that small granules undergo homotypic fusion to form larger granules, although the molecular and microscopic details of this theory are lacking. Ultimately, the immature granules further mature and become smaller by clathrin-coated vesicle-mediated removal of membrane and small amounts of digestive enzymes to the endosomal system resulting in concentration of the digestive enzyme content into a mature, electron-dense ZG78. AP1-mediated clathrin coat formation directed by MPRs also removes some, but not all, lysosomal enzymes from ISGs78.
Few studies have investigated the secretagogue regulation of post-Golgi zymogen granule formation. Kostenko et al. demonstrated a role for the tyrosine kinase c-src in mediating Golgi morphology and secretory granule formation86. Overexpression of c-src caused Golgi expansion whereas pharmacological inhibition reduced granule formation in cultured AR42J acinar cells and isolated acinar cells. These results provide the first known signaling pathway for acute Golgi-mediated ZG formation in response to secretory stimulation.
B. Anterograde Endosomal Trafficking through the Minor Secretory Compartment
The great majority of digestive enzyme secretion is mediated by ZG exocytosis at the apical membrane (Figures 2, 3). However, there are two additional and unique parallel secretory pathways identified in acinar cells from the pancreas and parotid glands known as the constitutive-like (CLP) and minor regulated (MRP) pathways as shown in Figure 387–92. Though these pathways only provide a small contribution to total protein secretion, they are likely important to acinar cell function. The CLP and MRP were identified by their rapid discharge (~2 h) of newly synthesized secretory proteins in pulse-chase studies, whereas ZG proteins were secreted by ~10 h under basal conditions88. Secretion from the CLP and MRP is acutely inhibited by brefeldin A (BFA), an inhibitor of guanine nucleotide exchange factors for class 1 ADP-ribosylation factors that function in vesicle formation from trans-Golgi and endosomal compartments87. The CLP and MRP were proposed to originate from vesicle fission at the TGN and ISG and traffic through an endosomal intermediate, subsequently identified as the early endosome prior to secretion at the apical membrane93–96. The MRP traffics from early or sorting endosomes directly to the apical membrane upon low-level secretagogue stimulation, whereas the CLP may subsequently enter an endosomal recycling compartment prior to exocytosis. More recent studies have identified that the endosomal, TGN peripheral membrane protein TPD52 and its associated proteins Rab5 and EEA1 play an important role in CLP trafficking in acinar cells96. It was also demonstrated that ZGs containing the SNARE protein vesicle-associated membrane protein 8 (VAMP8) require an intact endosomal pathway expressing D52, Rab5, and EEA1 in order to mature and/or undergo exocytosis97.
VI. CONCENTRATION OF NASCENT PROTEINS THROUGHOUT THE SECRETORY PATHWAY
Concentration of acinar cell nascent secretory proteins occurs throughout the pathway but is not uniform. Differences of over an order of magnitude have been observed for amylase, chymotrypsin, and procarboxypeptidase A, and occurs at multiple compartments in the secretory pathway63. The net effect of the enrichment mechanism is to concentrate soluble proteins such as amylase, trypsinogen, and chymotrypsinogen 10- to 20-fold between the ER lumen and the Golgi complex and even further enrichment as they move toward the zymogen granule and may be as high as a hundred-fold over the ER lumen. Though the mechanisms of such concentration remain unclear and likely vary among compartments, the decreasing pH gradient within the Golgi complex (going from pH 7.0 to 6.0) may contribute to this effect. Selection could also occur through interactions between secretory protein moieties and receptors; for example, the putative Golgi receptor muclin may interact with sulfated, O-glycosylated zymogens as a means to concentrate them in budding compartments98.
VII. SECRETORY GRANULE EXOCYTOSIS
Regulated secretion arises from a storage pool that excludes newly synthesized secretory proteins that accumulate during the intestinal interdigestive phase. It is able to release 15-30% of the gland’s secretory protein content by classical regulated exocytosis and is maximally stimulated following ingestion of a meal when release of massive amounts of digestive enzymes and zymogens (considering zymogens as a category of digestive enzyme) are required at rates greater than can be attained by protein synthesis alone. ZG exocytosis does not appear to contribute to resting secretion.
The purpose of these distinct mechanisms of secretion is unknown, but they likely serve to both ensure that some digestive enzymes will be present in the small intestine at all times and provide a secretory response that is proportional to luminal nutrients. A minor regulated pathway could serve to increase enzyme secretion in response to smaller quantities of food than presented by a full meal. Finally, a novel role for the constitutive and minor regulated compartments is that they might deliver the t-SNARES necessary for ZG fusion with the apical membrane99.
The release of ZG content into the lumen of the acinus requires fusion of the vesicular membrane with the apical plasma membrane. Four key steps are likely involved in this process: approximation of secretory granules in the apical region of the acinar cell, near the plasma membrane; tethering of secretory granules to the plasma membrane; docking and priming that involves SNARE proteins; and the final, calcium-dependent fusion event97.
The initial step, movement of the secretory granule from its site of formation in the trans-Golgi to the apical region of the cell, likely requires active involvement of contractile elements, particularly actin and associated motor proteins, in movement of the ZG to its apical plasma membrane target. It should be noted that in the resting interphase between rounds of exocytosis, an apical actin terminal web presumably negatively regulates resting secretion in that an actin mesh is always found between ZGs and the apical plasma membrane100–102. However, other actin roles have been proposed103. For example, an additional actin pool, regulated by the actin-polymerizing formin mDia1, mediates the final movement of zymogen granules to the apical membrane104. After reaching this most apical domain, the membrane of the ZG must recognize and become tethered to the apical plasma membrane prior to fusion. This implies that the actin meshwork beneath the apical membrane must be dissociated for close membrane apposition to occur, though additional steps, including overcoming fusion barriers, are needed before fusion can occur.
The role of microtubules in pancreatic acinar cell secretion is less clear. Microtubules are long, dynamic cytoskeletal structures composed of heterodimer polymers of α- and β-tubulin that undergo cycles of regulated polymerization and depolymerization105,106. During polymerization, β-subunits of one tubulin heterodimer contact the α-subunits of the next dimer resulting in one end of the microfilament having the α-subunits exposed and the other end with β-subunits exposed; these ends are designated the minus (−) and plus (+) ends, respectively. Microtubule motor proteins, such as kinesins and dynein, associate with select intracellular cargo and utilize ATP to facilitate transport along the microtubules107. Studies in the mid-1970s found that treatment of rodent pancreas both in vivo and ex vivo with the microtubule destabilizing compounds vinblastine and colchicine significantly inhibited, but did not fully prevent, secretagogue-stimulated amylase secretion108–111. Later work demonstrated that the minus ends of microtubules are anchored along the apical membrane and extend radially to the plus ends anchored in the basal cytoplasm112. Both kinesin and dynein have been identified in acini, though their localization (on ZGs, Golgi, ER) and effects on secretion are debated113–115. Interestingly, some studies show kinesin associating with ZGs in apical regions in response to secretory stimulation, an unexpected finding given that kinesins travel along microtubules in a minus-to-plus direction (i.e. anterograde in acini). Marlowe et al. speculate that organelles could contain both kinesin and dynein for bidirectional movement, and that kinesin could be involved in post-exocytosis membrane retrieval112. Additional high-resolution studies are needed to characterize the population and polarization of microtubules at the acinar apex to understand how kinesin shapes the secretory response.
The SNARE hypothesis for membrane recognition and fusion, which appears to be a generalized mechanism for all cells examined, is particularly relevant for the pancreatic acinar cell where specific interactions between the zymogen granule membrane and the apical plasma membrane ensure that exocytosis of digestive enzymes and proenzymes occurs into the acinar lumen116.
Two populations of ZGs have been identified in pancreatic acinar cells, those enriched in the SNARE protein VAMP2 and those enriched in Endobrevin/VAMP897. According to the SNARE hypothesis for exocytosis, VAMPs on the granule membrane interact with a syntaxin isoform and a SNAP isoform on the plasma membrane. Acinar cell apical plasma membrane contains syntaxins 2 and 4 and, interestingly, both apical plasma membrane and ZGs express SNAP3 and SNAP29117,118. Co-immunoprecipitation analysis revealed that VAMP2 ZGs interact with plasma membrane syntaxin 2 and SNAP23, whereas VAMP8 ZGs interact with apical membrane syntaxin 4 and SNAP23. ZG-ZG compound exocytosis which occurs during secretion was shown to involve VAMP8/syntaxin 3 and SNAP23 all of which are present on ZGs. A role for SNAP29 in acinar cell function has not been described.
To determine the roles of these VAMPs in the acinar cell, VAMP8 knockout mice together with adenoviral expression of tetanus toxin to selectively cleave VAMP2 were used to delineate the roles of VAMP2 versus VAMP8 ZG exocytosis during secretagogue stimulated secretion97. Results supported that VAMP2 and VAMP8 are the primary ZG SNAREs mediating stimulated but not basal secretion. Moreover, measuring acinar cell secretion over time in a perifusion apparatus revealed that VAMP2 ZG mediated an early immediate phase of secretion that peaks at two minutes and begins to decline followed by VAMP8 ZGs mediated second prolonged phase of secretion that peaks at five minutes and decays over 20 minutes. A subsequent study identified that the VAMP8, but not the VAMP2-mediated pathway, was primarily inhibited during high cholecystokinin (CCK)-induced acute acinar pancreatitis and that knockout of VAMP8 prevented most of the high dose CCK mediated secretory inhibition and fully blocked the accumulation of active trypsin in acinar cells119.
ZGs have been shown to undergo exocytosis at the basolateral plasma membrane during acute pancreatitis120. Evidence suggests that VAMP8 normally inhibits basolateral exocytosis; however PKC-mediated phosphorylation of the SNARE accessory protein Munc18c allows VAMP8 to mediate basolateral exocytosis in a SNARE complex involving VAMP8/syntaxin 4/SNAP23121. Presumably, release of digestive enzymes to the extracellular space enhances tissue damage thereby exacerbating disease progress.
A unique mechanism that has been observed in acinar cells is that of compound exocytosis, whereby secretory granule fuse with one another prior to interacting and fusing with the apical plasma membrane118,122. Though the physiological meaning of compound exocytosis is not currently understood, it may facilitate more rapid and efficient release of ZG content since the acinar lumen is a small fraction of the total surface area limiting the number of granules that can fuse with the apical membrane at any one time. What triggers granule to granule fusion prior to contact with the apical plasma membrane is unclear, but may involve changes in pH, changes in phospholipid content, acquisition of different SNARE proteins, etc., and needs further research.
VIII. REFERENCES
- Gemmer M, Förster F. A clearer picture of the ER translocon complex. 2020. J Cell Sci;133. PMID: 32019826.
- Akopian D, Shen K, Zhang X, Shan S. Signal recognition particle: an essential protein-targeting machine. Annu Rev Biochem 2013; 82:693–721. PMID: 23414305.
- Voss M, Schröder B, Fluhrer R. Mechanism, specificity, and physiology of signal peptide peptidase (SPP) and SPP-like proteases. Biochim Biophys Acta 2013; 1828:2828–2839. PMID: 24099004.
- Reinisch KM, De Camilli P. SMP-domain proteins at membrane contact sites: Structure and function. Biochim Biophys Acta 2016; 1861:924–927. PMID: 26686281.
- Son A, Park S, Shin DM, Muallem S. Orai1 and STIM1 in ER/PM junctions: roles in pancreatic cell function and dysfunction. Am J Physiol Cell Physiol 2016; 310:C414-422. PMID: 26739495.
- Nyathi Y, Wilkinson BM, Pool MR. Co-translational targeting and translocation of proteins to the endoplasmic reticulum. Biochim Biophys Acta 2013; 1833:2392–2402. PMID: 23481039.
- Waldron RT, Su H-Y, Piplani H, Capri J, Cohn W, Whitelegge JP, Faull KF, Sakkiah S, Abrol R, Yang W, Zhou B, Freeman MR, Pandol SJ, Lugea A. Ethanol Induced Disordering of Pancreatic Acinar Cell Endoplasmic Reticulum: An ER Stress/Defective Unfolded Protein Response Model. Cell Mol Gastroenterol Hepatol 2018; 5:479–497. PMID: 29930975.
- Grey MJ. Proteomic Study Defines How Alcohol Alters ER Structure and Redox Proteome to Trigger ER Stress and Acinar Cell Pathology in Pancreatitis. Cell Mol Gastroenterol Hepatol 2018; 5:640–641. PMID: 29713665.
- Desilva MG, Notkins AL, Lan MS. Molecular characterization of a pancreas-specific protein disulfide isomerase, PDIp. DNA Cell Biol 1997; 16:269–274. PMID: 9115635.
- Fu X-M, Dai X, Ding J, Zhu BT. Pancreas-specific protein disulfide isomerase has a cell type-specific expression in various mouse tissues and is absent in human pancreatic adenocarcinoma cells: implications for its functions. J Mol Histol 2009; 40:189–199. PMID: 19821078.
- Fujimoto T, Nakamura O, Saito M, Tsuru A, Matsumoto M, Kohno K, Inaba K, Kadokura H. Identification of the physiological substrates of PDIp, a pancreas-specific protein-disulfide isomerase family member. J Biol Chem 2018; 293:18421–18433. PMID: 30315102.
- Reily C, Stewart TJ, Renfrow MB, Novak J. Glycosylation in health and disease. Nat Rev Nephrol 2019; 15:346–366. PMID: 30858582.
- Wendorf P, Linder D, Sziegoleit A, Geyer R. Carbohydrate structure of human pancreatic elastase 1. Biochem J 1991; 278:505–514. PMID: 1898343.
- Abouakil N, Mas E, Bruneau N, Benajiba A, Lombardo D. Bile salt-dependent lipase biosynthesis in rat pancreatic AR 4-2 J cells. Essential requirement of N-linked oligosaccharide for secretion and expression of a fully active enzyme. J Biol Chem 1993; 268:25755–25763. PMID: 8245011.
- Bruneau N, Lombardo D. Chaperone Function of a Grp 94-related Protein for Folding and Transport of the Pancreatic Bile Salt-dependent Lipase. J Biol Chem 1995; 270:13524–13533. PMID: 7768954.
- Cooley MM, Thomas DDH, Deans K, Peng Y, Lugea A, Pandol SJ, Puglielli L, Groblewski GE. Deficient Endoplasmic Reticulum Acetyl-CoA Import in Pancreatic Acinar Cells Leads to Chronic Pancreatitis. Cell Mol Gastroenterol Hepatol 2020; Article "In Press"; S2352-345X(20):30171-5. PMID: 33080365.
- Peng Y, Li M, Clarkson BD, Pehar M, Lao PJ, Hillmer AT, Barnhart TE, Christian BT, Mitchell HA, Bendlin BB, Sandor M, Puglielli L. Deficient import of acetyl-CoA into the ER lumen causes neurodegeneration and propensity to infections, inflammation, and cancer. J Neurosci 2014; 34:6772–6789. PMID: 24828632.
- Pehar M, Lehnus M, Karst A, Puglielli L. Proteomic assessment shows that many endoplasmic reticulum (ER)-resident proteins are targeted by N(epsilon)-lysine acetylation in the lumen of the organelle and predicts broad biological impact. J Biol Chem 2012; 287:22436–22440. PMID: 22628546.
- Ye R, Mareninova OA, Barron E, Wang M, Hinton DR, Pandol SJ, Lee AS. Grp78 heterozygosity regulates chaperone balance in exocrine pancreas with differential response to cerulein-induced acute pancreatitis. Am J Pathol 2010; 177:2827–2836. PMID: 20971738.
- Kubisch CH, Logsdon CD. Endoplasmic reticulum stress and the pancreatic acinar cell. Expert Rev Gastroenterol Hepatol 2008; 2:249–260. PMID: 19072360.
- Waldron RT, Pandol S, Lugea A, Groblewski G. Endoplasmic Reticulum Stress and the Unfolded Protein Response in Exocrine Pancreas Physiology and Pancreatitis. Pancreapedia: The Exocrine Pancreas Knowledge Base. 2015. DOI: 10.3998/panc.2015.41.
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001; 107:881–891. PMID: 11779464.
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002; 415:92–96. PMID: 11780124.
- Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell 1999; 10:3787–3799. PMID: 10564271.
- Schindler AJ, Schekman R. In vitro reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc Natl Acad Sci USA 2009; 106:17775–17780. PMID: 19822759.
- Adachi Y, Yamamoto K, Okada T, Yoshida H, Harada A, Mori K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct Funct 2008; 33:75–89. PMID: 18360008.
- Bommiasamy H, Back SH, Fagone P, Lee K, Meshinchi S, Vink E, Sriburi R, Frank M, Jackowski S, Kaufman RJ, Brewer JW. ATF6alpha induces XBP1-independent expansion of the endoplasmic reticulum. J Cell Sci 2009; 122:1626–1636. PMID: 19420237.
- Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee A-H, Qian S-B, Zhao H, Yu X, Yang L, Tan BK, Rosenwald A, Hurt EM, Petroulakis E, Sonenberg N, Yewdell JW, Calame K, Glimcher LH, Staudt LM. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 2004; 21:81–93. PMID: 15345222.
- Lee A-H, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 2003; 23:7448–7459. PMID: 14559994.
- Lee A-H, Chu GC, Iwakoshi NN, Glimcher LH. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J 2005; 24:4368–4380. PMID: 16362047.
- Kowalik AS, Johnson CL, Chadi SA, Weston JY, Fazio EN, Pin CL. Mice lacking the transcription factor Mist1 exhibit an altered stress response and increased sensitivity to caerulein-induced pancreatitis. Am J Physiol Gastrointest Liver Physiol 2007; 292:G1123-1132. PMID: 17170023.
- Huh WJ, Esen E, Geahlen JH, Bredemeyer AJ, Lee A-H, Shi G, Konieczny SF, Glimcher LH, Mills JC. XBP1 controls maturation of gastric zymogenic cells by induction of MIST1 and expansion of the rough endoplasmic reticulum. Gastroenterology 2010; 139:2038–2049. PMID: 20816838.
- Tian X, Jin RU, Bredemeyer AJ, Oates EJ, Błażewska KM, McKenna CE, Mills JC. RAB26 and RAB3D Are Direct Transcriptional Targets of MIST1 That Regulate Exocrine Granule Maturation. Mol Cell Biol 2010; 30:1269–1284. PMID: 20038531.
- Reimold AM, Etkin A, Clauss I, Perkins A, Friend DS, Zhang J, Horton HF, Scott A, Orkin SH, Byrne MC, Grusby MJ, Glimcher LH. An essential role in liver development for transcription factor XBP-1. Genes Dev 2000; 14:152–157. PMID: 10652269.
- Lugea A, Tischler D, Nguyen J, Gong J, Gukovsky I, French SW, Gorelick FS, Pandol SJ. Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology 2011; 140:987–997. PMID: 21111739.
- Pehar M, Jonas MC, Hare TM, Puglielli L. SLC33A1/AT-1 protein regulates the induction of autophagy downstream of IRE1/XBP1 pathway. J Biol Chem 2012; 287:29921–29930. PMID: 22787145.
- Zhou L, Tan J, Cao R, Xu J, Chen X, Qi Z, Zhou S, Li S, Mo Q, Li Z, Zhang G. ATF6 regulates the development of chronic pancreatitis by inducing p53-mediated apoptosis. Cell Death Dis 2019; 10:1–12. PMID: 31506423.
- Iida K, Li Y, McGrath BC, Frank A, Cavener DR. PERK eIF2 alpha kinase is required to regulate the viability of the exocrine pancreas in mice. BMC Cell Biol 2007; 8:38. PMID: 17727724.
- Gao Y, Sartori DJ, Li C, Yu Q-C, Kushner JA, Simon MC, Diehl JA. PERK Is Required in the Adult Pancreas and Is Essential for Maintenance of Glucose Homeostasis. Mol Cell Biol 2012; 32:5129–5139. PMID: 23071091.
- Suyama K, Ohmuraya M, Hirota M, Ozaki N, Ida S, Endo M, Araki K, Gotoh T, Baba H, Yamamura K-I. C/EBP homologous protein is crucial for the acceleration of experimental pancreatitis. Biochem Biophys Res Commun 2008; 367:176–182. PMID: 18166146.
- Maurel M, Chevet E, Tavernier J, Gerlo S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem Sci 2014; 39:245–254. PMID: 24657016.
- Lipson KL, Ghosh R, Urano F. The role of IRE1alpha in the degradation of insulin mRNA in pancreatic beta-cells. PLoS One 2008; 3:e1648. PMID: 18286202.
- Bhattacharya A, Qi L. ER-associated degradation in health and disease – from substrate to organism. J Cell Sci 2019; 132, jcs.232850. PMID: 31792042.
- Sun S, Shi G, Han X, Francisco AB, Ji Y, Mendonça N, Liu X, Locasale JW, Simpson KW, Duhamel GE, Kersten S, Yates JR, Long Q, Qi L. Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc Natl Acad Sci USA 2014; 111:E582–E591. PMID: 24453213.
- Wilkinson S. Emerging Principles of Selective ER Autophagy. J Mol Biol 2020; 432:185–205. PMID: 31100386.
- Smith MD, Harley ME, Kemp AJ, Wills J, Lee M, Arends M, von Kriegsheim A, Behrends C, Wilkinson S. CCPG1 Is a Non-canonical Autophagy Cargo Receptor Essential for ER-Phagy and Pancreatic ER Proteostasis. Dev Cell 2018; 44:217-232.e11. PMID: 29290589.
- Smith MD, Wilkinson S. CCPG1, an unconventional cargo receptor for ER-phagy, maintains pancreatic acinar cell health. Mol Cell Oncol 2018; 5:e1441631. PMID: 30263939.
- Lahiri V, Klionsky DJ. CCPG1 is a noncanonical autophagy cargo receptor essential for reticulophagy and pancreatic ER proteostasis. Autophagy 2018; 14:1107–1109. PMID: 29911925.
- Farrugia MA, Puglielli L. Nε-lysine acetylation in the endoplasmic reticulum - a novel cellular mechanism that regulates proteostasis and autophagy. J Cell Sci 2018; 131:jcs.221747. PMID: 30446507.
- Barlowe CK, Miller EA. Secretory Protein Biogenesis and Traffic in the Early Secretory Pathway. Genetics 2013; 193:383–410. PMID: 23396477.
- Kurokawa K, Nakano A. The ER exit sites are specialized ER zones for the transport of cargo proteins from the ER to the Golgi apparatus. J Biochem 2019; 165:109–114. PMID: 30304445.
- Budnik A, Stephens DJ. ER exit sites--localization and control of COPII vesicle formation. FEBS Lett 2009; 583:3796–3803. PMID: 19850039.
- Bannykh SI, Rowe T, Balch WE. The organization of endoplasmic reticulum export complexes. J Cell Biol 1996; 135:19–35. PMID: 8858160.
- Jensen D, Schekman R. COPII-mediated vesicle formation at a glance. J Cell Sci 2011;124:1–4. PMID: 21172817.
- Matsuoka K, Schekman R, Orci L, Heuser JE. Surface structure of the COPII-coated vesicle. Proc Natl Acad Sci USA 2001; 98:13705–13709. PMID: 11717432.
- Appenzeller-Herzog C, Hauri H-P. The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. J Cell Sci 2006; 119:2173–2183. PMID: 16723730.
- Martínez-Menárguez JA, Geuze HJ, Slot JW, Klumperman J. Vesicular tubular clusters between the ER and Golgi mediate concentration of soluble secretory proteins by exclusion from COPI-coated vesicles. Cell 1999; 98:81–90. PMID: 10412983.
- Malkus P, Jiang F, Schekman R. Concentrative sorting of secretory cargo proteins into COPII-coated vesicles. J Cell Biol 2002; 159:915–921. PMID: 12499351.
- Khoriaty R, Everett L, Chase J, Zhu G, Hoenerhoff M, McKnight B, Vasievich MP, Zhang B, Tomberg K, Williams J, Maillard I, Ginsburg D. Pancreatic SEC23B deficiency is sufficient to explain the perinatal lethality of germline SEC23B deficiency in mice. Sci Rep 2016; 6:27802. PMID: 27297878.
- Khoriaty R, Vogel N, Hoenerhoff MJ, Sans MD, Zhu G, Everett L, Nelson B, Durairaj H, McKnight B, Zhang B, Ernst SA, Ginsburg D, Williams JA. SEC23B is required for pancreatic acinar cell function in adult mice. Mol Biol Cell 2017; 28:2146–2154. PMID: 28539403.
- Hauri HP, Kappeler F, Andersson H, Appenzeller C. ERGIC-53 and traffic in the secretory pathway. J Cell Sci 2000; 113:587–596. PMID: 10652252.
- Arakel EC, Schwappach B. Formation of COPI-coated vesicles at a glance. J Cell Sci 2018;131: jcs.209890. PMID: 29535154.
- Oprins A, Rabouille C, Posthuma G, Klumperman J, Geuze HJ, Slot JW. The ER to Golgi Interface is the Major Concentration Site of Secretory Proteins in the Exocrine Pancreatic Cell. Traffic 2001; 2:831–838. PMID: 11733050.
- Sesso A, de Faria FP, Iwamura ES, Corrêa H. A three-dimensional reconstruction study of the rough ER-Golgi interface in serial thin sections of the pancreatic acinar cell of the rat. J Cell Sci 1994; 107:517–528. PMID: 8006070.
- Glick BS, Luini A. Models for Golgi Traffic: A Critical Assessment. Cold Spring Harb Perspect Biol 2011; 3:a005215. PMID: 21875986.
- Papanikou E, Glick BS. Golgi compartmentation and identity. Curr Opin Cell Biol 2014;29:74–81. PMID: 24840895.
- Pantazopoulou A, Glick BS. A Kinetic View of Membrane Traffic Pathways Can Transcend the Classical View of Golgi Compartments. Front Cell Dev Biol 2019; 7:153. PMID: 31448274.
- Saraste J, Prydz K. A New Look at the Functional Organization of the Golgi Ribbon. Front Cell Dev Biol 2019; 7:171. PMID: 31497600.
- Klumperman J. Architecture of the mammalian Golgi. Cold Spring Harb Perspect Biol 2011; 3:a005181. PMID: 21502307.
- Rambourg A, Clermont Y, Hermo L. Formation of secretion granules in the Golgi apparatus of pancreatic acinar cells of the rat. Am J Anat 1988; 183:187–199. PMID: 2850745.
- McMahon HT, Mills IG. COP and clathrin-coated vesicle budding: different pathways, common approaches. Curr Opin Cell Biol 2004; 16:379–391. PMID: 1526170.
- Barois N, Bakke O. The adaptor protein AP-4 as a component of the clathrin coat machinery: a morphological study. Biochem J 2005; 385:503–510. PMID: 15377281.
- Bonifacino JS. The GGA proteins: adaptors on the move. Nat Rev Mol Cell Biol 2004; 5:23–32. PMID: 14708007.
- Slot JW, Geuze HJ. Immunoelectron microscopic exploration of the Golgi complex. J Histochem Cytochem 1983; 31:1049–1056. PMID: 6863900.
- Tokhtaeva E, Mareninova OA, Gukovskaya AS, Vagin O. Analysis of N- and O-Glycosylation of Lysosomal Glycoproteins. Methods Mol Biol 2017; 1594:35–42. PMID: 28456975.
- Wolters-Eisfeld G, Mercanoglu B, Hofmann BT, Wolpers T, Schnabel C, Harder S, Steffen P, Bachmann K, Steglich B, Schrader J, Gagliani N, Schlüter H, Güngör C, Izbicki JR, Wagener C, Bockhorn M. Loss of complex O-glycosylation impairs exocrine pancreatic function and induces MODY8-like diabetes in mice. Exp Mol Med 2018; 50:1–13. PMID: 30305605.
- Brown WJ, Farquhar MG. The mannose-6-phosphate receptor for lysosomal enzymes is concentrated in cis Golgi cisternae. Cell 1984; 36:295–307. PMID: 6319015.
- Klumperman J, Kuliawat R, Griffith JM, Geuze HJ, Arvan P. Mannose 6–Phosphate Receptors Are Sorted from Immature Secretory Granules via Adaptor Protein AP-1, Clathrin, and Syntaxin 6–positive Vesicles. J Cell Biol 1998; 141:359–371. PMID: 9548715.
- Braulke T, Bonifacino JS. Sorting of lysosomal proteins. Biochim Biophys Acta 2009; 1793:605–614. PMID: 19046998.
- Jamieson JD, Palade GE. Intracellular transport of secretory proteins in the pancreatic exocrine cell. I. Role of the peripheral elements of the Golgi complex. J Cell Biol 1967; 34:577–596. PMID: 6035647.
- Jamieson JD, Palade GE. Synthesis, intracellular transport, and discharge of secretory proteins in stimulated pancreatic exocrine cells. J Cell Biol 1971; 50:135–158. PMID: 4327462.
- Palade G. Intracellular aspects of the process of protein synthesis. Science 1975; 189:347–358. PMID: 1096303.
- Wendler F, Page L, Urbé S, Tooze SA. Homotypic Fusion of Immature Secretory Granules During Maturation Requires Syntaxin 6. Mol Biol Cell 2001; 12:1699–1709. PMID: 11408578.
- Ahras M, Otto GP, Tooze SA. Synaptotagmin IV is necessary for the maturation of secretory granules in PC12 cells. J Cell Biol 2006; 173:241–251. PMID: 16618809.
- Hammel I, Lagunoff D, Galli SJ. Regulation of secretory granule size by the precise generation and fusion of unit granules. J Cell Mol Med 2010; 14:1904–1916. PMID: 20406331.
- Kostenko S, Heu CC, Yaron JR, Singh G, de Oliveira C, Muller WJ, Singh VP. c-Src regulates cargo transit via the Golgi in pancreatic acinar cells. Sci Rep 2018; 8:11903. PMID: 30093675.
- von Zastrow M, Castle JD. Protein sorting among two distinct export pathways occurs from the content of maturing exocrine storage granules. J Cell Biol 1987; 105:2675–2684. PMID: 3500952.
- Arvan P, Castle JD. Phasic release of newly synthesized secretory proteins in the unstimulated rat exocrine pancreas. J Cell Biol 1987; 104:243–252. PMID: 2433293.
- Castle JD. Sorting and secretory pathways in exocrine cells. Am J Respir Cell Mol Biol 1990; 2:119–126. PMID: 2407275.
- Castle JD, Castle AM. Two regulated secretory pathways for newly synthesized parotid salivary proteins are distinguished by doses of secretagogues. J Cell Sci 1996; 109:2591–2599. PMID: 8923220.
- Hendricks LC, McClanahan SL, Palade GE, Farquhar MG. Brefeldin A affects early events but does not affect late events along the exocytic pathway in pancreatic acinar cells. Proc Natl Acad Sci USA 1992; 89:7242–7246. PMID: 1496018.
- Huang AY, Castle AM, Hinton BT, Castle JD. Resting (basal) secretion of proteins is provided by the minor regulated and constitutive-like pathways and not granule exocytosis in parotid acinar cells. J Biol Chem 2001; 276:22296–22306. PMID: 11301325.
- Thomas DDH, Kaspar KM, Taft WB, Weng N, Rodenkirch LA, Groblewski GE. Identification of Annexin VI as a Ca2+-sensitive CRHSP-28-binding Protein in Pancreatic Acinar Cells. J Biol Chem 2002; 277:35496–35502. PMID: 12105190.
- Thomas DDH, Weng N, Groblewski GE. Secretagogue-induced translocation of CRHSP-28 within an early apical endosomal compartment in acinar cells. Am J Physiol Gastrointest Liver Physiol 2004; 287:G253–G263. PMID: 14977633.
- Thomas DDH, Martin CL, Weng N, Byrne JA, Groblewski GE. Tumor protein D52 expression and Ca2+-dependent phosphorylation modulates lysosomal membrane protein trafficking to the plasma membrane. Am J Physiol Cell Physiol 2010; 298:C725–C739. PMID: 20032513.
- Messenger SW, Thomas DDH, Falkowski MA, Byrne JA, Gorelick FS, Groblewski GE. Tumor protein D52 controls trafficking of an apical endolysosomal secretory pathway in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol 2013;305:G439–G452. PMID: 23868405.
- Messenger SW, Falkowski MA, Thomas DDH, Jones EK, Hong W, Giasano HY, Boulis NM, Groblewski GE. Vesicle Associated Membrane Protein 8 (VAMP8)-mediated Zymogen Granule Exocytosis Is Dependent on Endosomal Trafficking via the Constitutive-Like Secretory Pathway. J Biol Chem 2014; 289:28040–28053. PMID: 25138214.
- Boulatnikov I, De Lisle RC. Binding of the Golgi sorting receptor muclin to pancreatic zymogens through sulfated O-linked oligosaccharides. J Biol Chem 2004; 279:40918–40926. PMID: 15292166.
- Castle AM, Huang AY, Castle JD. The minor regulated pathway, a rapid component of salivary secretion, may provide docking/fusion sites for granule exocytosis at the apical surface of acinar cells. J Cell Sci 2002; 115:2963–2973. PMID: 12082156.
- O’Konski MS, Pandol SJ. Effects of caerulein on the apical cytoskeleton of the pancreatic acinar cell. J Clin Invest 1990; 86:1649–1657. PMID: 1700797.
- Muallem S, Kwiatkowska K, Xu X, Yin HL. Actin filament disassembly is a sufficient final trigger for exocytosis in nonexcitable cells. J Cell Biol 1995; 128:589–598. PMID: 7860632.
- Valentijn KM, Gumkowski FD, Jamieson JD. The subapical actin cytoskeleton regulates secretion and membrane retrieval in pancreatic acinar cells. J Cell Sci 1999; 112:81–96. PMID: 9841906.
- Larina O, Bhat P, Pickett JA, Launikonis BS, Shah A, Kruger WA, Edwardson JM, Thorn P. Dynamic regulation of the large exocytotic fusion pore in pancreatic acinar cells. Mol Biol Cell 2007; 18:3502–3511. PMID: 17596517.
- Geron E, Schejter ED, Shilo B-Z. Directing exocrine secretory vesicles to the apical membrane by actin cables generated by the formin mDia1. Proc Natl Acad Sci USA 2013; 110:10652–10657. PMID: 23754409.
- Brouhard GJ, Rice LM. Microtubule dynamics: an interplay of biochemistry and mechanics. Nat Rev Mol Cell Biol 2018; 19:451–463. PMID: 29674711.
- Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer 2004; 4:253–265. PMID: 15057285.
- Caviston JP, Holzbaur ELF. Microtubule motors at the intersection of trafficking and transport. Trends Cell Biol 2006; 16:530–537. PMID: 16938456.
- Seybold J, Bieger W, Kern HF. Studies on intracellular transport of secretory proteins in the rat exocrine pancreas. II. Inhibition of antimicrotubular agents. Virchows Arch A Pathol Anat Histol 1975; 368:309–327. PMID: 813370.
- Williams JA, Lee M. Microtubules and pancreatic amylase release by mouse pancreas in vitro. J Cell Biol 1976; 71:795–806. PMID: 791957.
- Stock C, Launay JF, Grenier JF, Bauduin H. Pancreatic acinar cell changes induced by caerulein, vinblastine, deuterium oxide, and cytochalasin B in vitro. Lab Invest 1978; 38:157–164. PMID: 564425.
- Nevalainen TJ. Inhibition of pancreatic exocrine secretion by vinblastine. Res Exp Med (Berl) 1975; 165:163–168. PMID: 1224037.
- Marlowe KJ, Farshori P, Torgerson RR, Anderson KL, Miller LJ, McNiven MA. Changes in kinesin distribution and phosphorylation occur during regulated secretion in pancreatic acinar cells. Eur J Cell Biol 1998; 75:140–152. PMID: 9548371.
- Kraemer J, Schmitz F, Drenckhahn D. Cytoplasmic dynein and dynactin as likely candidates for microtubule-dependent apical targeting of pancreatic zymogen granules. Eur J Cell Biol 1999; 78:265–277. PMID: 10350215.
- Ueda N, Ohnishi H, Kanamaru C, Suzuki J, Tsuchida T, Mashima H, Yasuda H, Fujita T. Kinesin is involved in regulation of rat pancreatic amylase secretion. Gastroenterology 2000; 119:1123–1131. PMID: 11040199.
- Schnekenburger J, Weber I-A, Hahn D, Buchwalow I, Krüger B, Albrecht E, Domschke W, Lerch MM. The role of kinesin, dynein and microtubules in pancreatic secretion. Cell Mol Life Sci 2009; 66:2525–2537. PMID: 19488676.
- Rothman JE. The principle of membrane fusion in the cell (Nobel lecture). Angew Chem Int Ed Engl 2014; 53:12676–12694. PMID: 25087728.
- Falkowski MA, Thomas DDH, Messenger SW, Martin TF, Groblewski GE. Expression, localization, and functional role for synaptotagmins in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol 2011; 301:G306-316. PMID: 21636530.
- Behrendorff N, Dolai S, Hong W, Gaisano HY, Thorn P. Vesicle-associated Membrane Protein 8 (VAMP8) Is a SNARE (Soluble N-Ethylmaleimide-sensitive Factor Attachment Protein Receptor) Selectively Required for Sequential Granule-to-granule Fusion. J Biol Chem 2011; 286:29627–29634. PMID: 21733851.
- Messenger SW, Jones EK, Holthaus CL, Thomas DDH, Cooley MM, Byrne JA, Mareninova OA, Gukovskaya AS, Groblewski GE. Acute acinar pancreatitis blocks vesicle-associated membrane protein 8 (VAMP8)-dependent secretion, resulting in intracellular trypsin accumulation. J Biol Chem 2017; 292:7828–7839. PMID: 28242757.
- Gaisano HY, Gorelick FS. New Insights Into the Mechanisms of Pancreatitis. Gastroenterology 2009; 136:2040–2044. PMID: 19379751.
- Lam PPL, Cosen Binker LI, Lugea A, Pandol SJ, Gaisano HY. Alcohol redirects CCK-mediated apical exocytosis to the acinar basolateral membrane in alcoholic pancreatitis. Traffic 2007; 8:605–617. PMID: 17451559.
- Wäsle B, Turvey M, Larina O, Thorn P, Skepper J, Morton AJ, Edwardson JM. Syncollin is required for efficient zymogen granule exocytosis. Biochem J 2005; 385:721–727. PMID: 15462671.