
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2016.6
| Attachment | Size |
|---|---|
| 658 KB |
1. Introduction
The natural history of chronic pancreatitis (CP) includes progressive loss of exocrine and endocrine function. Endocrine failure occurs due to progressive destruction of the gland by the ongoing inflammatory events of CP, and results in diabetes which is termed pancreatogenic or type 3c diabetes. The pathophysiology of type 3c diabetes (T3cDM) includes loss of secretion of the principal glucoregulatory hormones produced by the islets (insulin, glucagon, and pancreatic polypeptide), and is contributed to by abnormal secretion of the incretin hormones glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) which are adversely affected by the loss of exocrine function. T3cDM is therefore a complex form of secondary diabetes, which requires careful assessment and management.
2. Pathophysiology of T3cDM
Definition and Classification of Pancreatogenic or Type 3c Diabetes
Diabetes caused by agenesis, destruction or loss of the exocrine pancreas has been termed Pancreatogenic Diabetes. In 1979, the National Diabetes Data Group (NDDG) of the National Institutes of Health formulated a classification system that was subsequently endorsed by the American Diabetes Association (ADA) and other groups which defined secondary diabetes due to a variety of causes including pancreatic disease as a third type of diabetes, after type 1 and type 2 (24). In 1997, the ADA published a table of diabetes classification in which pancreatogenic diabetes was designated as type 3c diabetes (T3cDM) (39). The ADA classification table was subsequently republished as an annual supplement every year from 2002 (19) until 2014 (4). In 2015, the same classification table was not re-published, and pancreatogenic diabetes was included as a form of “diabetes due to other causes” without further description or guidelines for diagnosis or therapy (3). The term type 3c diabetes has been used repeatedly by investigators and clinicians to refer to the diabetes which results from or is associated with pancreatitis, trauma/pancreatectomy, neoplasia, cystic fibrosis, hemochromatosis, and fibrocalcific pancreatopathy.
It is uncertain whether pancreatogenic diabetes due to CP is identical to other forms of T3cDM due to causes such as pancreatectomy, pancreatic cancer, or cystic fibrosis, or whether these various causes of T3cDM have similar etiologies. T3cDM due to CP appears to most closely resemble the form of diabetes associated with pancreatic resection, in that beta-cell mass is reduced according to the extent of the resection or disease, and a deficiency of pancreatic polypeptide (PP), a regulator of the expression and availability of hepatic insulin receptors, is a consequence of advanced CP as well as proximal (or total) pancreatectomy (15). In diabetes that is a consequence, and frequently a harbinger, of pancreatic ductal adenocarcinoma (PDAC), insulin secretion is impaired due to what is thought to be a para-neoplastic phenomenon (29), and insulin sensitivity is impaired (11). The impairment in (hepatic) insulin sensitivity may be a consequence of the loss of PP secretion associated with PDAC localized to the pancreatic head (27). In diabetes associated with cystic fibrosis, the progressive destruction of the pancreas due to the impairment in bicarbonate secretion caused by mutations in the cystic fibrosis transmembrane receptor is associated with insulin deficiency as well as a loss of hepatic insulin sensitivity (30) which may also be a consequence of impaired PP secretion (1).
Prevalence of T3cDM
The prevalence of pancreatogenic or T3cDM was believed to be quite low until recent studies by Hardt et al (25) and Ewald et al (18) showed that T3cDM accounted for 8-9% of the total population of over 1900 diabetic patients referred to an academic center in Germany (Figure 1). No such prevalence study has been published for North America or elsewhere, due to uncertainly of the criteria for the designation of T3cDM and its differentiation from type 1 and type 2 diabetes. In the German series, it was found that half of the patients with probable or definite T3cDM had been previously misdiagnosed as having either type 1 diabetes (T1DM) or type 2 diabetes (T2DM). The largest fraction (76%) of patients classified as having T3cDM had antecedent CP.
Natural History of CP-associated T3cDM
The prevalence of diabetes in patients with CP has been reported to range from 20-70% (8, 44). Longitudinal studies of 500 patients with (alcohol-induced) CP showed that after 25 years, 83% of patients had developed diabetes, and most required insulin treatment (36). The morbidity of the diabetes due to small vessel disease (retinopathy and nephropathy) has been shown to be similar for T3cDM as for other types (14), and the mortality of T3cDM due to hypoglycemia and other diabetic complications appears comparable of that of diabetes due to other causes.

Figure 1. Distribution of types of diabetes (a) and causes of type 3c (pancreatogenic) diabetes (b) based on studies of 1,922 diabetic patients referred to an academic medical center. From Ref (15), based on data in Ref (25), with permission.
Pathophysiology of T3cDM
Severe T3cDM secondary to a complete loss of islet hormone secretion is associated with a form of disease termed “brittle diabetes” due to a loss of both insulin and glucagon secretion. Insulin deficiency results in an increase in peripheral insulin sensitivity, and the potential for over-medication despite small doses of insulin, and glucagon deficiency results in a loss of hypoglycemia awareness and responsiveness with resulting neuroglycopenia. A 1981 series of 117 patients treated with partial and total pancreatectomy for CP revealed that half of the late deaths after surgery were due to hypoglycemia (21).
Hyperglycemia in T3cDM is due to insulin deficiency, and persistent endogenous glucose production due to the loss of hepatic insulin sensitivity (15). In laboratory and clinical studies, persistent hepatic glucose production was seen to be associated with the loss of PP secretion (47, 49), and was reversed by PP administration in PP-deficient patients with CP (10).
Progressive exocrine dysfunction results in a failure to digest fats, due to lipase deficiency. This results in impaired absortion of the fat-soluble vitamins A, D, E, and K (2), and is associated with a high prevalence of metabolic bone disease and fracture in CP (51). The loss of fat digestion also results in impaired incretin-mediated insulin release due to altered GLP-1 and GIP secretion from the proximal and distal small bowel. Pancreatic enzyme replacement therapy (PERT) has been shown to restore incretin secretion and improve nutrient-induced insulin release (16, 31). A comparison of the clinical and laboratory features which differentiate T1DM, T2DM and T3cDM is shown in Table 1.


Differentiation of T3cDM from T1DM and T2DM
The diagnosis of T3cDM due to CP was addressed in a consensus conference held at the annual meeting of PancreasFest in 2012 (46). A definition of major and minor criteria for the diagnosis of T3cDM was described, and included the documentation of antecedent pancreatic exocrine disease established by radiology and exocrine secretory testing. In addition, the absence of anti-islet antibodies (to rule out T1DM) and PP deficiency (to rule out T2DM) were included as criteria. These criteria have also been used by Ewald et al. in their studies of T3cDM (17). As the majority of patients with CP will develop diabetes later in their disease course, the most common misdiagnosis is that of T2DM. A proposed set of criteria for the diagnosis of T3cDM is shown in Table 2.
PP secretion is increased in obesity associated with diabetes (23), and with normal aging and age-related diabetes (12). PP secretion is increased in these diabetic groups presumably as a compensatory response to a progressive decline in insulin sensitivity. In T3cDM due to CP, pancreatic resection, pancreatic carcinoma, and cystic fibrosis, however, PP secretion is impaired (15) (Table 1). Therefore, a failure of PP secretion in response to oral nutrients is a useful test to discriminate T3cDM from the more prevalent T2DM. PP secretion is stimulated by glucose, protein and fat, but glucose alone is a relatively weak stimulant for PP release. This is probably a consequence of the normal enteric stimulation of PP release by cholecystokinin (CCK) (33) and GIP (5, 12). Because oral glucose is a weak stimulant of PP, it is recommended that a mixed meal test be used to document PP levels (46). Eight ounces of a liquid nutritional supplement such as Boost®or Ensure® serves as a suitable nutrient stimulus for PP release and is usually well tolerated. PP levels normally increase 3- to 5-fold within 30-60 minutes after ingestion of the liquid meal, so plasma levels obtained before (fasting) and at 30 and 60 minutes after ingestion are sufficient to detect a failure (less than 2-fold increase) in PP secretion.
Practical Importance of Differentiating T3cDM from T2DM
T3cDM differs from T2DM due to the high prevalence of metabolic bone disease and nutritional deficiencies, and an increased risk of pancreatic cancer. Pancreatic exocrine insufficiency is present in most patients with T3cDM, and frequently exists despite the absence of the classic symptom of steatorrhea. When T3cDM is diagnosed or suspected, it is appropriate to assess exocrine function in all patients, and the most commonly used test is the fecal elastase 1 (FE1) level (17). Levels of FE1 above 200ug/g are considered normal, whereas levels below 100ug/g indicate significant exocrine impairment (34). Low FE1 levels or a history suggestive of exocrine insufficiency are indications for PERT.
Metabolic bone disease, due to a loss of vitamin D absorption, is common in patients with CP and T3cDM. Vitamin D supplements along with PERT are believed to be useful to reduce the risk of osteopenia and bone fractures (2), and should be considered in all patients.
CP associated with diabetes carries a 12- to 33-fold increased risk for the development of pancreatic cancer (9, 32, 35). Therefore all patients with T3cDM due to CP should be regularly evaluated for the presence of this malignancy. Indications of the presence of PDAC include unexplained weight loss, or a sudden worsening of glycemic control in a patient with known diabetes. Surveillance studies might include CA19-9 levels, although this has not been shown to be a useful marker of early stage (resectable) PDAC. No published criteria have yet been formalized for the screening of CP patients, other than periodic pancreatic imaging. Although CT scanning is used most widely, endoscopic ultrasound is more useful for the detection of early-stage disease. Patients with stable CP who suddenly develop diabetes are candidates for pancreatic imaging studies to rule out the possibility of PDAC.

3. Management of T3cDM
Screening for Diabetes
All patients with CP should be periodically evaluated for the presence of the development of T3cDM (46). Surveillance should include hemoglobin A1C (Hgb A1C)levels or fasting glucose levels, as recommended by the ADA (Table 3). Values which are suspicious or non-diagnostic for diabetes should be followed by oral glucose tolerance testing (OGTT) to confirm the presence of diabetes. Repeat testing should be performed every 6 months if equivocal or non-diagnostic, or every 3 years if normal (3). Differentiation of T3cDM from T2DM should include the criteria shown in Table 2 with the measurement of PP responsiveness to a liquid test meal, as described above.
PP deficiency may be the earliest indication of endocrine dysfunction in CP. Because PP-secreting cells are localized predominantly in the pancreatic head and uncinate process, inflammation localized to the pancreatic head may affect PP secretion before global beta-cell failure results in hyperglycemia. PP deficiency is associated with impaired hepatic insulin sensitivity, but may not result in diabetes if sufficient residual islet function is present (47).
Algorithm for the Management of Hyperglycemia
The goal of therapy is to lower the HgbA1C level to less than 7%. No evidence-based recommendations are available to guide the treatment of T3cDM, specifically. Therefore guidelines which pertain to the management of T2DM are usually followed (42). Oral therapy is recommended to begin with metformin, although this drug may be problematic to patients with CP, due to gastrointestinal irritability. A schedule of graduated increases in metformin doses has been recommended to lessen side effects (Table 4) and maybe used with periodic HgbA1C testing to assess the effectiveness of therapy.

If metformin is not tolerated, or is ineffective, additional or alternative medications include alpha glucosidase inhibitors (αGIs) (45), thiazolidinediones (TZDs) (22), and sodium-glucose cotransporter-2 inhibitors (SGLT2Is) (40). TZDs have been shown in one study to be effective in reversing the hepatic insulin resistance of T3cDM due to CP (52), but SGLT2Is have been associated with euglycemic acidosis in insulin deficient patients (28) so their use should be accompanied by caution.
Insulin secretagogues and incretin-based therapy should be avoided or delayed when possible, due to the increased incidence of hypoglycemia associated with sulfonylurea therapy, and the suspected (but unconfirmed) risk of pancreatitis and pancreatic malignancy associated with incretin-based therapy. It is recommended that until studies of incretin-based therapy are found safe in patients with T3cDM, that these agents be avoided (46). In CP patients with impaired glucagon secretion, the risk of hypoglycemia is increased. Metformin, TZDs, and αGIs have not been shown to increase the risk of hypoglycemia compared to other therapies (7). Patients with a history of hypoglycemia might better be managed with a therapeutic goal of maintaining HgbA1C level to less than 8%.
When hyperglycemic crises occur, or when HgbA1C levels are persistently above 7%, insulin treatment is indicated for the management of T3cDM (15). Patients with T3cDM frequently require low doses of insulin, due to increased peripheral insulin sensitivity, and a usual approach is to begin with 10 units of Lantus insulin once per day with subsequent assessment of HgbA1C levels and surveillance for hypoglycemia. Additional doses of insulin may be necessary as twice-a-day administration or greater. Patients with brittle diabetes may be better managed with a programmable insulin pump coupled with a continuous glucose monitor.
Importance of Pancreatic Enzyme Replacement Therapy (PERT)
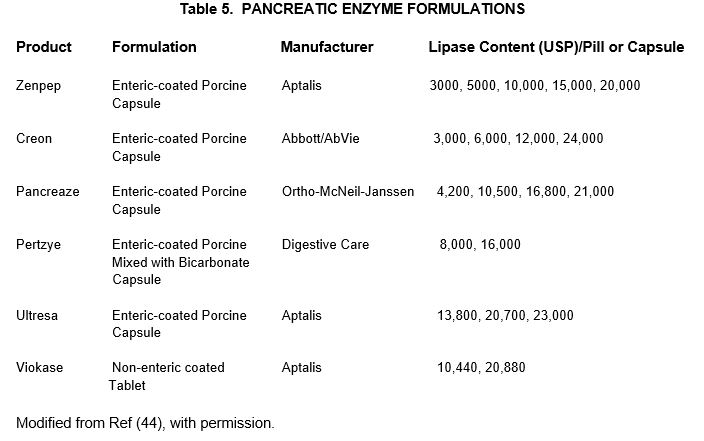
Pancreatic exocrine insufficiency is present in virtually all patients with T3cDM, and may not be accompanied by the classic symptom of steatorrhea (26), so PERT is recommended for consideration in all cases. Oral enzyme supplements vary in formulation and dosage, and their availability is affected by healthcare coverage and distribution issues. Therefore, multiple formulations of pancreatic enzyme supplements are potentially useful, with selection based on availability (Table 5).
In general, 90,000 USP units of enzyme are required for complete digestion and absorption of a normal meal. The amount of enzymes required by an individual patient will depend on residual endogenous exocrine function, and may be limited by side effects. It is recommended that therapy begin with a dose of 50,000-60,000 USP units, or 1,000 USP units of lipase/kg, per meal, taken in divided doses before and after eating, with subsequent assessment of effectiveness and symptoms (2). Pancreatic enzyme bioavailability is affected by acid inactivation of lipase, so an antacid medication is usually prescribed to improve efficacy (20). Multivitamins or vitamin D supplements are also appropriate to consider in patients with T3cDM due to the high prevalence of metabolic bone disease in CP.
Role of Early Intervention in Delaying or Preventing Progressive Endocrine Failure
Progressive endocrine dysfunction is a sign of progressive pancreatic destruction due to CP. Therefore therapeutic interventions which halt or delay the continued inflammation may prevent or delay the development of T3cDM. These interventions may include therapeutic endoscopy or surgical approaches to prevent persistent inflammation, in addition to abstinence from toxic agents (e.g., alcohol and nicotine) which are associated with recurrent attacks. The relief of obstructive pancreatopathy by surgical decompression has been shown to prevent or delay the progression of CP over 24 months (43), although the development of subsequent T3cDM after resectional and hybrid surgical procedures has been found to be progressive and similar after 5 years (48). The risk of T3cDM after surgical procedures is less in the near-term post-operative period with hybrid procedures (e.g., Beger or Frey procedures) than with proximal pancreatectomy (6).
Role of Total Pancreatectomy with Islet Auto-transplantation (TPIAT) in Preserving Endocrine Function in CP
The principal symptom which usually prompts treatment of CP is pain. Most patients with recurrent or chronic pain due to CP can be successfully managed with therapeutic endoscopic or surgical resection and/or decompressive approaches. A significant number of patients have persistent symptoms despite prior treatment approaches, and/or are not considered appropriate candidates for decompressive or hybrid procedures. Many of these patients are disabled by their symptoms, and are usually dependent on opioid treatment for relief. For this subset of CP patients who still have endocrine function, an alternative consideration for management is TPIAT.
Total pancreatectomy (alone) is a potentially devastating procedure because of the risk of complete exocrine and endocrine insufficiency. Although such patients can be managed with meticulous attention to nutritional and glycemic homestasis, the requirements of this care are very high. In some patients with disabling symptoms, total pancreatectomy may be a feasible option, if endocrine function can be preserved by autologous islet transplantation. TPIAT has been performed since 1977, when it was developed at the University of Minnesota (41). Currently, there are 15 centers in the United States which offer TPIAT, with 150-200 such procedures being performed each year. About 30% of TPIAT patients are insulin-independent 3 years after their procedure, another 32% have partial islet function, and more than 85% of patients report significant pain relief and an improved quality of life after recovery (50). In recent years, TPIAT has been employed in an increasing number of pediatric patients with hereditary or idiopathic CP, whose disease is often completely disabling. In children aged 5-12 years who have received TPIAT, 56% of patients are insulin-independent and virtually all have returned to normal activities one year after the procedure (13). In adolescent patients aged 13-19 years, 41% of patients are insulin-independent and 90% report relief of symptoms and a return to normal activities 3 years after the procedure.
4. Summary
Endocrine failure is a common complication of CP, and indicates severe or worsening disease. The development of T3cDM may be hastened by pancreatic resection or pre-existing diminished pancreatic reserve. T3cDM is a result of the combined deficiency of insulin, glucagon, and PP, and is worsened by an impaired incretin effect, due to exocrine deficiency-related deficits in the secretion of GLP-1 and GIP. T3cDM is virtually always accompanied by exocrine insufficiency, which results in vitamin D deficiency and metabolic bone disease. Pancreatic enzyme replacement is therefore indicated in almost all cases. The management of T3cDM requires careful attention to the risks of hypoglycemia, and should begin with a trial of therapy with metformin. New onset T3cDM is an indication to consider the possible presence of pancreatic cancer as a cause. Reducing the risk of T3cDM through interventions which delay or prevent its occurrence are important considerations in the treatment of CP.
5. References
- Adrian TE, McKiernan J, Johnstone DI, Hiller EJ, Vyas H, Sarson DL, et al. Hormonal abnormalities of the pancreas and gut in cystic fibrosis. Gastroenterology 79(3): 460-465, 1980. PMID: 7000612.
- Afghani E, Sinha A and Singh VK. An overview of the diagnosis and management of nutrition in chronic pancreatitis. Nutr Clin Pract 29(3): 295-311, 2014. PMID: 24743046.
- American Diabetes Association. (2) Classification and diagnosis of diabetes. Diabetes Care 38 Suppl: S8-S16, 2015. PMID: 25537714.
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 37 Suppl 1: S81-90, 2014. PMID: 24357215.
- Amland PF, Jorde R, Aanderud S, Burhol PG and Giercksky KE. Effects of intravenously infused porcine GIP on serum insulin, plasma C-peptide, and pancreatic polypeptide in non-insulin-dependent diabetes in the fasting state. Scand J Gastroenterol 20(3): 315-320, 1985. PMID: 3890139.
- Andersen DK and Frey CF. The evolution of the surgical treatment of chronic pancreatitis. Ann Surg 251(1): 18-32, 2010. PMID: 20009754.
- Anderson M, Powell J, Campbell KM and Taylor JR. Optimal management of type 2 diabetes in patients with increased risk of hypoglycemia. Diabetes Metab Syndr Obes 7: 85-94, 2014. PMID: 24623984.
- Bank S. Chronic pancreatitis: clinical features and medical management. Am J Gastroenterol 81(3): 153-167, 1986. PMID: 3513542.
- Brodovicz KG, Kou TD, Alexander CM, O'Neill EA, Engel SS, Girman CJ, et al. Impact of diabetes duration and chronic pancreatitis on the association between type 2 diabetes and pancreatic cancer risk. Diabetes Obes Metab 14(12): 1123-1128, 2012. PMID: 22831166.
- Brunicardi FC, Chaiken RL, Ryan AS, Seymour NE, Hoffmann JA, Lebovitz HE, et al. Pancreatic polypeptide administration improves abnormal glucose metabolism in patients with chronic pancreatitis. J Clin Endocrinol Metab 81(10): 3566-3572, 1996. PMID: 8855802.
- Cersosimo E, Pisters PW, Pesola G, McDermott K, Bajorunas D and Brennan MF. Insulin secretion and action in patients with pancreatic cancer. Cancer 67(2): 486-493, 1991. PMID: 1985741.
- Chia CW, Odetunde JO, Kim W, Carlson OD, Ferrucci L and Egan JM. GIP contributes to islet trihormonal abnormalities in type 2 diabetes. J Clin Endocrinol Metab 99(7): 2477-2485, 2014. PMID: 24712564.
- Chinnakotla S, Bellin MD, Schwarzenberg SJ, Radosevich DM, Cook M, Dunn TB, et al. Total pancreatectomy and islet autotransplantation in children for chronic pancreatitis: indication, surgical techniques, postoperative management, and long-term outcomes. Ann Surg 260(1): 56-64, 2014. PMID: 24509206.
- Couet C, Genton P, Pointel JP, Louis J, Gross P, Saudax E, et al. The prevalence of retinopathy is similar in diabetes mellitus secondary to chronic pancreatitis with or without pancreatectomy and in idiopathic diabetes mellitus. Diabetes Care 8(4): 323-328, 1985. PMID: 4042797.
- Cui Y and Andersen DK. Pancreatogenic diabetes: special considerations for management. Pancreatology 11(3): 279-294, 2011. PMID: 21757968.
- Ebert R and Creutzfeldt W. Reversal of impaired GIP and insulin secretion in patients with pancreatogenic steatorrhea following enzyme substitution. Diabetologia 19(3): 198-204, 1980. PMID: 6997121.
- Ewald N and Bretzel RG. Diabetes mellitus secondary to pancreatic diseases (Type 3c)--are we neglecting an important disease? Eur J Intern Med 24(3): 203-206, 2013. PMID: 23375619.
- Ewald N, Kaufmann C, Raspe A, Kloer HU, Bretzel RG and Hardt PD. Prevalence of diabetes mellitus secondary to pancreatic diseases (type 3c). Diabetes Metab Res Rev 28(4): 338-342, 2012. PMID: 22121010.
- Expert Committee on the Diagonsis and Classification of Diabetes Mellitus. Report of the expert committee on the diagnosis and classification of diabetes mellitus. Diabetes Care 26 Suppl 1: S5-20, 2003. PMID: 12502614.
- Forsmark CE. Management of chronic pancreatitis. Gastroenterology 144(6): 1282-1291 e1283, 2013. PMID: 23622138.
- Gall FP, Muhe E and Gebhardt C. Results of partial and total pancreaticoduodenectomy in 117 patients with chronic pancreatitis. World J Surg 5(2): 269-275, 1981. PMID: 7245796.
- Garber AJ, Abrahamson MJ, Barzilay JI, Blonde L, Bloomgarden ZT, Bush MA, et al. AACE comprehensive diabetes management algorithm 2013. Endocr Pract 19(2): 327-336, 2013. PMID: 23598536.
- Glaser B, Zoghlin G, Pienta K and Vinik AI. Pancreatic polypeptide response to secretin in obesity: effects of glucose intolerance. Horm Metab Res 20(5): 288-292, 1988. PMID: 3042579.
- Group NDD. Classification and diagnosis of diabetes mellitus and other categories of glucose intolerance. Diabetes 28(12): 1039-1057, 1979. PMID: 510803.
- Hardt PD, Brendel MD, Kloer HU and Bretzel RG. Is pancreatic diabetes (type 3c diabetes) underdiagnosed and misdiagnosed? Diabetes Care 31 Suppl 2: S165-169, 2008. PMID: 18227480.
- Hardt PD, Hauenschild A, Nalop J, Marzeion AM, Jaeger C, Teichmann J, et al. High prevalence of exocrine pancreatic insufficiency in diabetes mellitus. A multicenter study screening fecal elastase 1 concentrations in 1,021 diabetic patients. Pancreatology 3(5): 395-402, 2003. PMID: 14526149.
- Hart PA, Baichoo E, Bi Y, Hinton A, Kudva YC and Chari ST. Pancreatic polypeptide response to a mixed meal is blunted in pancreatic head cancer associated with diabetes mellitus. Pancreatology 15(2): 162-166, 2015. PMID: 25766398.
- Hine J, Paterson H, Abrol E, Russell-Jones D and Herring R. SGLT inhibition and euglycaemic diabetic ketoacidosis. Lancet Diabetes Endocrinol 3(7): 503-504, 2015. PMID: 26025388.
- Javeed N, Sagar G, Dutta SK, Smyrk TC, Lau JS, Bhattacharya S, et al. Pancreatic Cancer-Derived Exosomes Cause Paraneoplastic beta-cell Dysfunction. Clin Cancer Res 21(7): 1722-1733, 2015. PMID: 25355928.
- Kien CL, Horswill CA, Zipf WB, McCoy KS and O'Dorisio T. Elevated hepatic glucose production in children with cystic fibrosis. Pediatr Res 37(5): 600-605, 1995. PMID: 7603777.
- Knop FK, Vilsboll T, Larsen S, Hojberg PV, Volund A, Madsbad S, et al. Increased postprandial responses of GLP-1 and GIP in patients with chronic pancreatitis and steatorrhea following pancreatic enzyme substitution. Am J Physiol Endocrinol Metab 292(1): E324-330, 2007. PMID: 16954337.
- Liao KF, Lai SW, Li CI and Chen WC. Diabetes mellitus correlates with increased risk of pancreatic cancer: a population-based cohort study in Taiwan. J Gastroenterol Hepatol 27(4): 709-713, 2012. PMID: 21929650.
- Lonovics J, Guzman S, Devitt P, Hejtmancik KE, Suddith RL, Rayford PL, et al. Release of pancreatic polypeptide in humans by infusion of cholecystokinin. Gastroenterology 79(5 Pt 1): 817-822, 1980. PMID: 7419006.
- Loser C, Mollgaard A and Folsch UR. Faecal elastase 1: a novel, highly sensitive, and specific tubeless pancreatic function test. Gut 39(4): 580-586, 1996. PMID: 8944569.
- Maisonneuve P, Lowenfels AB, Bueno-de-Mesquita HB, Ghadirian P, Baghurst PA, Zatonski WA, et al. Past medical history and pancreatic cancer risk: Results from a multicenter case-control study. Ann Epidemiol 20(2): 92-98, 2010. PMID: 20123159.
- Malka D, Hammel P, Sauvanet A, Rufat P, O'Toole D, Bardet P, et al. Risk factors for diabetes mellitus in chronic pancreatitis. Gastroenterology 119(5): 1324-1332, 2000. PMID: 11054391.
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF and Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28(7): 412-419, 1985. PMID: 3899825.
- Meier JJ, Menge BA, Breuer TG, Muller CA, Tannapfel A, Uhl W, et al. Functional assessment of pancreatic beta-cell area in humans. Diabetes 58(7): 1595-1603, 2009. PMID: 19509022.
- Mellitus TECotDaCoD. Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 20(7): 1183-1197, 1997. PMID: 9203460.
- Nair S and Wilding JP. Sodium glucose cotransporter 2 inhibitors as a new treatment for diabetes mellitus. J Clin Endocrinol Metab 95(1): 34-42, 2010. PMID: 19892839.
- Najarian JS, Sutherland DE, Baumgartner D, Burke B, Rynasiewicz JJ, Matas AJ, et al. Total or near total pancreatectomy and islet autotransplantation for treatment of chronic pancreatitis. Ann Surg 192(4): 526-542, 1980. PMID: 6775603.
- Nathan DM, Buse JB, Davidson MB, Ferrannini E, Holman RR, Sherwin R, et al. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 32(1): 193-203, 2009. PMID: 18945920.
- Nealon WH and Thompson JC. Progressive loss of pancreatic function in chronic pancreatitis is delayed by main pancreatic duct decompression. A longitudinal prospective analysis of the modified puestow procedure. Ann Surg 217(5): 458-466; discussion 466-458, 1993. PMID: 8489308.
- Nyboe Andersen B, Krarup T, Thorsgaard Pedersen NT, Faber OK, Hagen C and Worning H. B cell function in patients with chronic pancreatitis and its relation to exocrine pancreatic function. Diabetologia 23(2): 86-89, 1982. PMID: 6182047.
- Riccardi G, Giacco R, Parillo M, Turco S, Rivellese AA, Ventura MR, et al. Efficacy and safety of acarbose in the treatment of Type 1 diabetes mellitus: a placebo-controlled, double-blind, multicentre study. Diabet Med 16(3): 228-232, 1999. PMID: 10227568.
- Rickels MR, Bellin M, Toledo FG, Robertson RP, Andersen DK, Chari ST, et al. Detection, evaluation and treatment of diabetes mellitus in chronic pancreatitis: recommendations from PancreasFest 2012. Pancreatology 13(4): 336-342, 2013. PMID: 23890130.
- Seymour NE, Brunicardi FC, Chaiken RL, Lebovitz HE, Chance RE, Gingerich RL, et al. Reversal of abnormal glucose production after pancreatic resection by pancreatic polypeptide administration in man. Surgery 104(2): 119-129, 1988. PMID: 3041640.
- Strate T, Bachmann K, Busch P, Mann O, Schneider C, Bruhn JP, et al. Resection vs drainage in treatment of chronic pancreatitis: long-term results of a randomized trial. Gastroenterology 134(5): 1406-1411, 2008. PMID: 18471517.
- Sun YS, Brunicardi FC, Druck P, Walfisch S, Berlin SA, Chance RE, et al. Reversal of abnormal glucose metabolism in chronic pancreatitis by administration of pancreatic polypeptide. Am J Surg 151(1): 130-140, 1986. PMID: 3946744.
- Sutherland DE, Radosevich DM, Bellin MD, Hering BJ, Beilman GJ, Dunn TB, et al. Total pancreatectomy and islet autotransplantation for chronic pancreatitis. J Am Coll Surg 214(4): 409-424; discussion 424-406, 2012. PMID: 22397977.
- Tignor AS, Wu BU, Whitlock TL, Lopez R, Repas K, Banks PA, et al. High prevalence of low-trauma fracture in chronic pancreatitis. Am J Gastroenterol 105(12): 2680-2686, 2010. PMID: 20736937.
- Zhou X and You S. Rosiglitazone inhibits hepatic insulin resistance induced by chronic pancreatitis and IKK-beta/NF-kappaB expression in liver. Pancreas 43(8): 1291-1298, 2014. PMID: 25036911.