
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2015.41
1. Endoplasmic Reticulum Stress and the Unfolded Protein Response
The endoplasmic reticulum (ER) plays a pivotal role in cellular homeostasis as it is the site of translation, folding and covalent modification of proteins either destined for secretion or expressed on the surface of other organelles and the plasma membrane. The cytoplasmic surface of the ER membrane is also the primary site of triglyceride, phospholipid and sterol synthesis and therefore, is a hub of membrane synthesis and cellular metabolism. Many studies have established the ER as a major site of intracellular Ca2+ storage essential for hormone/neurotransmitter signaling. More recent work has also begun to identify unexpected functions of the ER in diverse cellular processes including mitochondrial fission, endosomal dynamics and autophagy.
Quality control mechanisms for protein folding in the ER were first demonstrated by the identification of glucose-regulated proteins (GRP) 78 (a.k.a. BiP) and 94 (a.k.a. HSP90B1/endoplasmin) that reside in the ER lumen (34). Whereas GRP have significant homology to heat-shock chaperone proteins, glucose starvation, sulfhydryl reducing agents, inhibitors of glycosylation, and calcium ionophores, rather than heat stress, induce GRP expression. A seminal study demonstrated that enhanced expression of misfolded proteins in the ER induced GRP mRNA expression, thereby establishing the presence of a signaling network from the ER to the nucleus (28). The concept of an unfolded protein response (UPR) opened the door to a multitude of studies over the last 27 years uncovering molecular components and mechanisms of the UPR. This has provided important insight into the pathophysiological consequences of ER stress in various disorders from neurodegenerative to metabolic diseases. Perhaps not surprisingly given the high protein secretory capacity and vastly expanded ER in the pancreatic acinar cell, studies support that the UPR is critical to maintaining acinar cell homeostasis. This review will focus on what is known of the UPR in acinar cell function as well as recent evidence underscoring its importance in the development and progression of pancreatitis.
2. The UPR is Comprised of Distinct Functional Components
The UPR is activated when the concentration of unfolded or misfolded proteins present in the ER overwhelms the capacity of ER-resident proteins, including molecular chaperones, disulfide isomerases, oxidoreductases, acetylases and glycosylases, to respond to that stress.
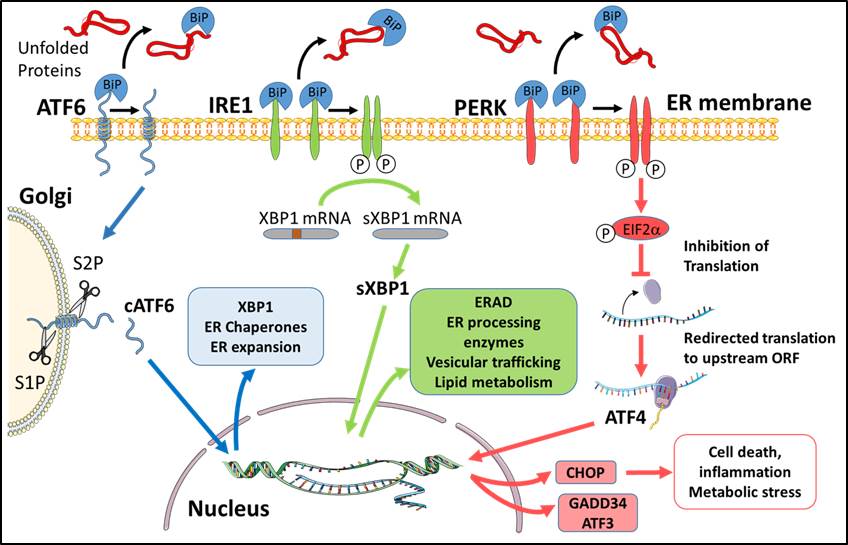
Figure 1. Principal components of the UPR/ER-stress response. ATF6, IRE1 and PERK are all ER transmembrane proteins capable of sensing the level of unfolded or misfolded proteins in the ER lumen. The precise mechanism(s) for activating each pathway is unclear and likely involves the displacement of molecular chaperones e.g. BiP from the luminal domain of the protein. ATF6: Upon activation, ATF6 traffics from the ER to the Golgi via COPII-directed clathrin coated vesicles. In the Golgi, ATF6 is sequentially cleaved by the site-1 and site-2 proteases freeing the N-terminal transcription factor domain (cATF6) to translocate to the nucleus and promote transcription of UPR target genes. IRE1: Activation of IRE1 kinase activity is mediated by trans-autophosphorylation causing IRE1 to oligomerize in the ER membrane. Phosphorylated IRE1 exhibits endoribonuclease activity that excises a portion of the XBP1 mRNA forming a spliced mRNA that generates the transcription factor sXBP1. sXBP1 translocates to the nucleus and directs the expression of a number of ER-regulators necessary for protein folding, lipid metabolism, vesicular trafficking and acinar secretory function. PERK: Kinase activation and autophosphorylation of PERK phosphorylates the cytosolic eukaryotic translation initiation factor eIF2α thereby inhibiting global secretory protein translation. As a consequence, translation of select mRNAs including ATF4 is redirected to an upstream open reading frame giving rise to alternative protein products. ATF4 acts as a transcriptional activator or repressor, and directs the synthesis of CHOP, also a transcription factor, that plays a key role in mediating cell death, inflammation and metabolic stress.
Figure 1 illustrates many of the features discussed in this and subsequent sections. Three major pathways of the UPR have been identified; the inositol-requiring enzyme-1 (IRE1), activating transcription factor 6 (ATF6), and protein kinase RNA (PKR)-like ER kinase (PERK) pathways. These transmembrane proteins can sense the level of stress in the ER lumen, and transduce signals to the cytoplasm; complex sensing mechanisms involve the binding of either unfolded proteins themselves, or chaperones, e.g., BiP that modulate their oligomerization and other individual functional responses. The level and duration of ER stresses dictate the extent to which each UPR pathway becomes activated. Acutely, new protein synthesis is attenuated to limit the protein load entering the ER. At later times, nuclear signaling induces gene expression to promote protein folding as well as degradation of misfolded proteins. Under extreme conditions of ER stress intensity and duration, UPR responses mitigating ER stress are supplanted by those promoting cell death.
The IRE1 and XBP1/spliced XBP1 (sXBP1) Pathway
IRE1 was originally identified in yeast as a gene required to enhance the expression of KAR2 (known as Binding immunoglobulin Protein (BiP) or 78 kDa glucose-regulated protein (GRP-78) in mammalian cells) during ER stress (12, 47). The name inositol-requiring enzyme-1 was coined based on its role in inositol prototropy in yeast (48). There are two isoforms, IRE1α and IRE1β, the latter of which is prominent in intestinal epithelia and its function is unclear. This review will focus on IRE1α, i.e., IRE1. IRE1 is a type 1 transmembrane protein with an N-terminal domain in the ER lumen that binds to BiP and a C-terminal serine/threonine protein kinase domain oriented to the cytoplasm. Displacement of the molecular chaperone BiP from the N-terminus and/or a direct interaction with unfolded proteins has been proposed to activate IRE1 kinase activity. Although kinase activity is required for signaling, the only known substrate characterized is IRE1 itself, which upon trans-autophosphorylation causes IRE1 to oligomerize in the ER membrane (12, 47, 65). Phosphorylated IRE1 induces an endoribonuclease activity in its C-terminus that excises a portion of the mRNA encoding the transcription factor homologous to ATF/CREB1 (HAC) in yeast (44) or X-box binding protein-1 (XBP1) in mammals (10, 78). Spliced XBP1 induces the expression of a number of ER proteins to mitigate ER stress (see below). With high levels of, or prolonged ER stress, IRE1 also participates in a process termed “regulated IRE1-dependent decay” (RIDD) of ER membrane-bound and cytosolic mRNAs as well as IRE1 mRNA to reduce protein synthesis (25, 26). More recently, IRE1 was also reported to cleave microRNAs that control the level of caspase-2 thereby promoting apoptosis (69). It is proposed that IRE1-mediated XBP1 splicing has a prosurvival output, whereas RIDD has a proapoptotic output (44). A final consideration is that IRE1 may serve as a link between ER stress and stress kinase activation. IRE1 recruits an adapter protein, TRAF2, to the ER membrane to initiate a signaling cascade that culminates in activation of Jun N-terminal Kinase (JNK) (70). Although JNK is a stress kinase that can induce apoptosis, the significance of this activation during physiological ER stress is unclear.
Activated IRE1 mediates the splicing of an intron in XBP1 mRNA that causes a frame-shift during translation, resulting in a new carboxyl terminal domain in the spliced XBP1 (sXBP1) protein. Unspliced XBP1 (MW 29-33 kDa) has a nuclear exclusion signal and rapidly degrades but can act as a negative modulator of the UPR when accumulated in mammalian cells (79). Conversely, sXBP1 (54-60 kDa) contains a nuclear localization signal and basic leucine-zipper (BZIP) domain that binds to the cis-acting ER stress response element (ERSE) and functions as a potent transcription factor. Genetic profiling and analyses revealed that sXBP1 controls the expression of genes related to the UPR including chaperone induction, up-regulation of ER-associated degradation (ERAD) machinery, membrane biogenesis, and ER quality control (33, 62, 64). In mammals, sXBP1 also activates the expression of cell type-specific targets linked to cell differentiation, signaling, and DNA damage (1, 33). In pancreatic acinar cells sXBP1 has been shown to be essential to maintain a differentiated secretory phenotype (32). This role of sXBP1 was demonstrated using pharmacologic inhibitors of IRE1 endonuclease activity in the acinar cell line, AR42J (13). Thus, IRE1 inhibition substantially diminished the spontaneous amylase secretion by these cells. We also find that the inhibition of the formation of sXBP1 attenuates the synthesis of certain digestive enzymes by acinar cells, as well as secretagogue-induced secretion. (unpublished data) That the efficacy of these approaches relies on inhibition of splicing reaction indicates that the spliced form of XBP1 plays a major role in biosynthesis and secretion in professional secretory cells. Nevertheless, unspliced XBP1 too may yet prove to act in unsuspected ways to alter acinar cell physiology. Interestingly, the unspliced form was reported to regulate the stability of sXBP1, and more recently a role was also proposed for unspliced XBP1 to regulate autophagy via control of the stability of another transcription factor, Foxo1 (72).
The PERK/ATF4 pathway
The protein kinase RNA (PKR)-like ER kinase (PERK) is a type 1 transmembrane protein that, like IRE1, undergoes kinase activation in response to accumulation of misfolded proteins in the ER lumen. Autophosphorylated PERK in turn phosphorylates the cytosolic eukaryotic translation initiation factor eIF2α at Ser51 causing its inactivation. Inactivation of eIF2α, a small G-protein, involves blocking its interaction with a guanine nucleotide exchange factor thereby locking it in a GDP bound state. This greatly reduces protein translation and acutely attenuates secretory protein load (23). Besides PERK, other kinases including general control nonderepressible 2 (GCN2), protein kinase RNA-activated (PKR) and heme-regulated inhibitor (HRI) can promote phosphorylation of eIF2α at Ser51 in response to cellular stresses such as amino acid deprivation, viral infection and heme deficiency.
In addition to inhibiting general protein translation, PERK activation also increases the translation of mRNAs that contain upstream open reading frames within their 5’UTR including the transcription factor ATF4 (22). Under stress conditions and eIF2α inhibition, translation is redirected to the upstream open reading frame giving rise to an alternative protein product. Only translation from the upstream open reading frame gives rise to a stable form of ATF4. ATF4 is a BZIP transcription factor of the cAMP response element binding protein family that forms homomeric and heteromeric dimers with various proteins and can act as a transcriptional activator or repressor. Analysis of transcripts from ATF4 knockout cells support that it regulates a number of genes that are important for secretory cell function (17). However, the best characterized ATF4 gene targets include CHOP (transcriptional factor C/EBP homologous protein), GADD34 (growth arrest and DNA damage-inducible 34), and activating transcription factor 3 (ATF3). CHOP was reported to play a major role in promoting apoptosis by inducing the expression of pro-apoptotic proteins including Bim, and more recently, DR5 and repressing the expression of anti-apoptotic proteins, e.g. Bcl2, as reviewed in (36, 45, 49, 54, 68). An alternate theory put forward by some investigators emphasizes enhanced expression and function of translational machinery to de-energize the cell by consuming excessive amounts of metabolic intermediates, rather than differential induction of pro- and suppression of anti-apoptotic proteins, as a major role of CHOP (43, 73). Irrespective of the precise pathway, CHOP induction correlates with cell death and knockout models have established its role as a detrimental signal, including in the exocrine pancreas.
A theme in the regulatory control of UPR is feedback within and/or between the different branches. CHOP (and ATF4) reportedly induces expression of GADD34, a regulatory subunit of protein phosphatase 1. Thus, GADD34 enables a stress-inducible phosphatase that dephosphorylates eIF2α producing negative feedback to inactivate its own induction (since eif2α Ser51 phosphorylation mediates CHOP expression). Although GADD34 can help to restore protein translation, it also promotes the expression of pro-apoptotic proteins, as evidenced by knockout as well as pharmacologic approaches (73). ATF3 is also a BZIP transcription factor of the cAMP response element binding protein family. Expression of ATF3 can be induced by a number of stresses in addition to the UPR and has been shown to have both positive and negative effects on cell survival and disease progression (21). Recently ATF3 was shown to interact with an acetyltransferase, Tat-interactive protein 60 (Tip60) that acetylates the major DNA damage kinase Ataxia telangiectasia mutated (ATM) to promote genomic stability during cell stress (14).
The ATF6 pathway
Two ubiquitously expressed isoforms, ATF6α and ATF6β are found in mammalian cells. Although they share significant homology, ER localization and processing, studies in cultured mouse embryonic fibroblasts from double knockout mice indicate ATF6α and not ATF6β is required for transcriptional induction of genes (2). Activated ATF6 mediates expansion of the ER and induces the expression of chaperones, foldases, and components of the ERAD pathway (2, 8). ATF6 is a type II ER transmembrane protein which like IRE1 and PERK has a C-terminal luminal domain but differs in that it has a BZIP transcription factor domain in the N-terminal cytoplasmic domain. During ER stress, and potentially via the loss of BiP binding, ATF6 emerges from the ER on COPII coated vesicles and traffics to the Golgi where it is sequentially cleaved by the site-1 and site-2 proteases (S1P and S2P) (60). Once released, the N-terminal transcription factor domain (cATF6) translocates to the nucleus and activates transcription of UPR target genes (24, 77).
3. ER Stress, UPR and Functional Cross-Talk in Pancreatitis Models
Synthesis and packaging of proteins for transport is the singular key task of the acinar cell of the exocrine pancreas. Accordingly, the ER of the acinar cell is highly developed; and all of the components of the UPR as described above have been identified in the acinar cell. The importance of the UPR to exocrine pancreatic function was demonstrated by ablating the XBP1 gene in mice, expressing an XBP1 transgene in liver to rescue them from embryonic lethality (32). These mice have normal morphology of all organs except the exocrine pancreas and salivary glands and die shortly after birth due to poor ER development and digestive enzyme synthesis in the acinar cells resulting in severe exocrine insufficiency (32). We have confirmed these results by a mouse with acinar cell specific conditional knock down of XBP1 (unpublished data). XBP1 deficiency leads to extensive acinar cell loss and severe pathology in the remaining acinar cells, as evidenced by a poorly developed ER network and secretory system, reduced expression of ER chaperones, marked reduction in zymogen granules and digestive enzymes, and accumulation of autophagic vacuoles.
The activation of UPR pathways has been demonstrated in several models of experimental pancreatitis (5, 16, 27, 29-31, 35, 41, 42, 59, 61, 67, 75, 76, 81). In the arginine-induced and high-dose cholecystokinin (CCK) models there was phosphorylation of PERK and its downstream target eIF2α, ATF4 translocation into the nucleus and increased CHOP expression accompanied by upregulation of BiP and sXBP1 (29, 31). Under these conditions, there was development of pancreatitis with inflammation and trypsin activation. On the other hand, secretagogues that do not cause pancreatitis, such as bombesin and the CCK analogue JMV-180, caused only increases in BiP and sXBP1 (30, 41). The chemical chaperones, tauroursodeoxycholic acid and 4-phenyl butyric acid reduced these markers of ER stress associated with a reduction in the severity of pancreatitis (41, 61). Finally, altering the expression of BiP has an effect on the severity of pancreatitis (76). UPR pathways are involved not only in acute pancreatitis models but also in chronic pancreatitis models (59).
Our understanding of the role of specific components of the UPR system in physiology and disease remains incomplete. Nevertheless, there are key findings that provide a framework for further investigations. One key finding is that genetic deletion of CHOP results in less severe experimental pancreatitis, suggesting that activation of the PERK-eIF2α pathway leading to increased CHOP expression is important for the development of the pathologic pancreatitis response (67). Indeed, Chop-/- mice exhibited less pancreatic inflammation and histological damage than wild type when challenged with cerulein and LPS (67).
We showed that the expression of sXBP1 increases in the pancreas of alcohol-fed animals. Further, we observed in these alcohol fed animals that pathologic changes of pancreatitis occur with genetic inhibition of the sXBP1 response to alcohol feeding (37, 38, 50). With inhibition of the sXBP1 response, there was activation of the PERK/eIF2α/ATF4/CHOP pathway associated with the development of pancreatitis. That is, we found that depriving the acinar cell from using the IRE1-sXBP1 to adapt to ER stress is associated with sustained activation of the PERK/eIF2α/ATF4/CHOP pathway and development of pancreatic pathology (37). From these findings we postulate that the IRE1/sXBP1 pathway is activated with mild to moderate ER stressors such as alcohol abuse; and that pancreas pathology develops when this ER “adaptive” response fails to reestablish ER homeostasis, reinforcing activation of the PERK/eIF2α/ATF4/CHOP pathways, inflammation and cell death. These findings also align with our concept that UPR mediates necessary adaptive responses to stressors to maintain normal function of the exocrine pancreas (illustrated in the above case with alcohol); and that cellular failure and pancreatitis ensue when the stressors exceed the adaptive capacity of the cell; or the adaptive response fails to deploy. We anticipate that completion of ongoing proteomic analysis of ER from XBP1 deficient mice will clarify which key proteins are altered, as well as how the deficiencies are pathologically exacerbated by risk factors such as ethanol consumption.
The findings and hypothesis listed above are consistent with results in other systems showing that CHOP promotes inflammation by regulating cytokine production, inflammatory and cell survival (19, 40). Our studies representing pathologic ER stress mechanisms arising in the exocrine pancreas also complement those attributed to beta cell malfunction in the endocrine pancreas that reportedly contribute to diabetic pathology (73). Indeed, the links between endocrine and exocrine dysfunction have been insufficiently elucidated despite many years of study; there is increasing awareness that each of these compartments participates reciprocally in the regulation of pancreas damage and disease.
ER stress, ERAD and Autophagy
ER stress is also linked with activation of ERAD (endoplasmic reticulum associated degradation) and induction of autophagic gene expression, mechanisms to eliminate misfolded proteins and dysfunctional ER (9, 80). These misfolded proteins are degraded by the proteasome in conjunction with autophagy to disassemble proteins and recycle their amino acids. Misfolded proteins within the ER are recognized and targeted for ERAD by mechanisms utilizing ER chaperones and quality control systems. Several ERAD regulators are regulated by sXBP1 and both cells and mice deficient in sXBP1 exhibit impaired ERAD and activation of autophagy (37, 38). We reported that Xbp+/- mice fed alcohol diets exhibited reduced pancreatic EDEM1, a key ERAD protein. This decrease was associated with marked vacuolization suggesting that a failure of the adaptive IRE1-sXBP1 pathway inhibits ERAD and promotes disordered autophagy (37, 38, 50).
ThePERK-eIF2α arm of the UPR induces autophagy through downstream targets including ATF4 and CHOP. Autophagy regulators required for induction and autophagosome formation, including REDD1, ATG3, ATG5, ATG7 and LC3B, as well as p62 are transcriptionally up regulated by ATF4 and CHOP (4, 57). These findings highlight the linkage between ER stress and induction of autophagy, two important contributors to the pathology of pancreatitis.
The Roles of ER Ca2+, Oxidative Stress and Other Factors in ER stress/UPR
The ER is one of the major storage sites for Ca2+ used to generate cytosolic signals to trigger diverse events including secretion and mitosis in all mammalian cells (7). The role of inositol lipid turnover in Ca2+ release from ER was originally demonstrated in pancreatic acinar cells (66). Extensive studies established the roles of both intracellular and extracellular Ca2+ in regulated secretion of zymogens from acinar cells (18). Localized Ca2+ spikes are required for optimal secretagogue-induced amylase release, while global cytosolic Ca2+ elevations in the acinar cell have been associated with supraphysiological inhibition of secretion and aberrant conversion of trypsinogen to active trypsin. Whereas these events represent widely accepted root causes of initial damage to acinar cells, NF-kB activation and an inflammatory response are also required for full pancreatitis development (58). Ca2+ stored within ER has important functions in protein biosynthesis and folding. Membrane-bound (calnexin) or lumenal (calreticulin) lectins, are major Ca2+-binding proteins within ER (74). These “glycosensor” proteins recycle cargo until its core glycosylation meets a quality standard for packaging and export from the ER en route to the Golgi. Ca2+ also regulates the ATPase activity of BiP that, in turn regulates its interactions including with unfolded proteins (6).
Whether Ca2+ affects the ER stress sensor functions through regulation of specific BiP interaction sites or independently, depletion of Ca2+ from ER is well established as a major inducer of ER stress. Thapsigargin (TG), an irreversible Ca2+ pump inhibitor, potently induces cellular XBP1 splicing, as well as PERK/eif2α/CHOP and ATF6 pathways. Administration of TG to cells elicits cell type-specific effects ranging from cell growth arrest to apoptosis. The ER stress associated with TG treatment is caused by perturbation of multiple functions, including protein synthesis, folding and formation of cargo-laden transport vesicles. To counterbalance this plethora of defects, XBP1s governs several measures that reduce the load of misfolded proteins, either through chaperone-assisted cycles of redox-dependent refolding, or removal via ERAD.
ER Ca2+ depletion and redox alterations are causally linked. Increases in misfolded proteins upregulate the activity and/or gene expression of the folding oxidoreductase, ERO-1L that generates H2O2 as it restores the balance of oxidized to reduced protein disulfide isomerases. In ongoing studies, we are examining effects of ethanol to perturb the redox status of key proteins of the folding and quality control apparatus as well as the various cargo molecules (zymogens) in transit to the secretory pathway. The scheme shown in Fig. 2 illustrates aspects of our working hypothesis for these studies.
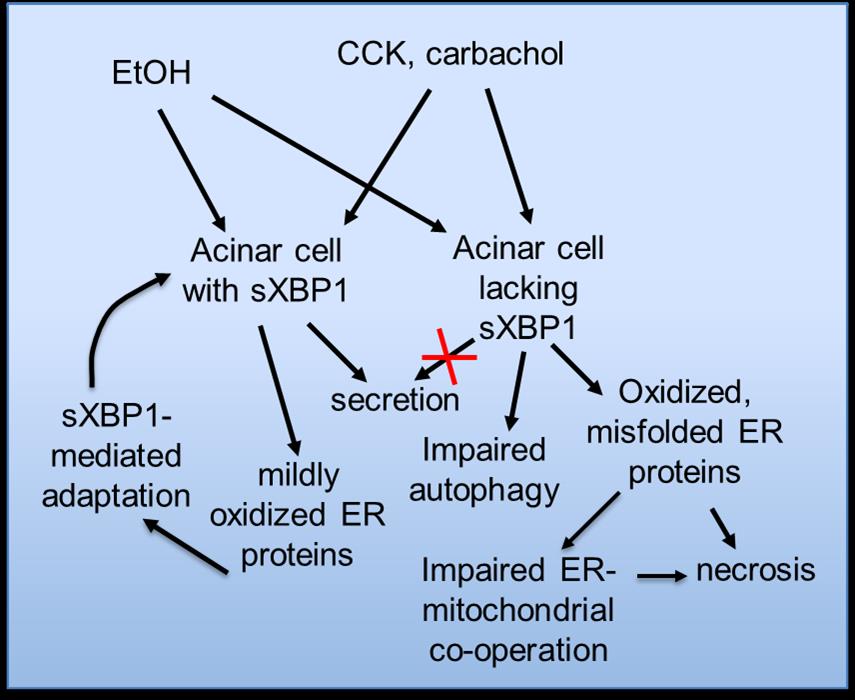
Figure 2. Scheme depicting UPR activation and regulation of ER stress responses. Working hypothesis for adaptation versus pathological outcomes to insults triggering redox perturbations in acinar cells in the presence or absence of the protein unfolded response regulator, spliced XBP1 (sXBP1).
Importantly, the ER stores a major part, but not all of the cellular Ca2+ used for signaling. Endo-lysosomal “acidic” organelles have also increasingly been implicated in physiological events (18, 51). In particular, specialized Ca2+-ATPases (SERCA3), and release channels (two pore channels and Mcoln1/TRPML1) have been associated with the storage and release events, which have been linked to lysosomal functions and autophagy in other cell types (46, 71). Pharmacologic agents that have helped to identify these stores in pancreatic acinar cells include glycyl-L-phenylalanine-beta-naphthylamide (GPN), bafilomycin and nigericin. Recently it was reported that all these agents release Ca2+ from both ER and endo-lysosomal Ca2+ stores. Much evidence points to cooperative domains, especially the membranes of distinct organelles, such as mitochondrial, lysosomal or plasma membrane in apposition to ER. Whereas the most essential among these has not been identified, it seems clear that the multiple Ca2+ stores of ER and other membrane-bound organelles operate interdependently (56). Recent studies of membrane contact sites uncovered specific complexes, e.g., EGFR-PTP1B, Rab7-VAP (via ORP1L or RILP), and STARD3-VAP that tether segments of ER with endosomes, and Nvj1p-Vac8p that mediates nuclear-vacuole tethering (3, 15, 52, 55).
Reversible Transcriptional Regulation of Acinar Cell Secretory Differentiation: Compensatory Regrowth after Damage versus Tumorigenesis
A highly specialized transcription factor of the helix-loop-helix (HLH) family, termed BHLHA15 or MIST1, was shown to dramatically alter the fate of the ER and secretory pathways. The expression of MIST1 is reportedly regulated by XBP1s (1). Konieczny and others established that proper pancreatic acinar cell organization and secretory function were MIST1 dependent by a knockout approach (53). Subsequently, Mist1-/- mice proved an interesting model to study the role of Ca2+ signaling in the acinar cell. The altered balance of ER versus secretory compartments revealed their roles in Ca2+ transients and global cytosolic elevations (39). Further studies probing the roles of distinct transcriptional pathways in altering the identity of acinar cells continue to reveal the unexpected phenotypic plasticity of this extraordinary cell type.
Mounting evidence indicates that acinar cells can give rise to tumors as well as to regenerative self-duplication, both of which occur through an initial loss of acinar cell properties and transition to a dedifferentiated phenotype. In this process, acinar genes such as carboxypeptidase 1 (CPA1) and pancreatic amylase (Amy2) are lost and expression of some genes important for embryonic development or conferring stem like properties, including Sox-9, Pdx1, Ptf-1 and intermediates of the Wnt/β-catenin signaling pathway are recapitulated. Dedifferentiation is permissive for proliferation, and in regeneration after damage, is followed by restoration of the acinar phenotype. In contrast, further phenotypic transition, i.e., acinar-to-ductal metaplasia (ADM) formation may occur, in parallel with or leading to pancreatic intraepithelial neoplasia (PanIN) lesions. PanIN represent definitive precursor lesions for pancreatic ductal adenocarcinoma (PDAC), whereas ADM are only tentatively established as such. Which of these (regrowth or tumorigenic) pathways is followed depends in part on environmental factors, including the presence or absence of an inflammatory milieu.
Interestingly, the Map kinases, extracellular signal-regulated kinase 1/2 (ERK1/2) and the BZIP transcription factor c-jun, are required for dedifferentiation (11, 20). Much evidence also supports a role for ERK-dependent signaling as a critical effector through which KRas activity promotes PanIN and PDAC formation. MIST1 expression restrains these activities of KRas and promotes maintenance of the acinar phenotype. Specifically, removing MIST1 was a permissive event for formation of ADM (63). Mechanisms whereby Ca2+, redox and UPR pathways impact MIST1 or other transcription factors to modulate phenotypic plasticity remain relatively unexplored. Elucidation of these pathways enable harnessing the replicative potential of dedifferentiated cells and reengineering acinar and/or endocrine cells “in reverse” to achieve novel forms of stem cell therapy.
4. Summary
ER stress and oxidative stress are interlocked stresses regulated by Ca2+ and interchanged via intimate membrane contacts in the pancreatic acinar cell. Distinct UPR pathways work to maintain the acinar secretory phenotype and restrain oncogenic signaling (sXBP1, MIST1) and others (CHOP) are proapoptotic. Multiple investigations support the view of sXBP1 mediated signaling as an adaptive branch of UPR that protects/maintains the functional phenotype of the pancreatic acinar cell. Prolonged ER stress impairs homeostatic processes such as endo/lysosomal function and autophagy (see Fig. 2). Collectively, such disturbances in the harmonious function of the acinar cell lead to pathologic outcomes. In particular, the energetic imbalance and oxidative stress that accompany excessive unfolded ER proteins and impaired ER-mitochondrial cooperation promote necrosis, a form of cell death associated with inflammatory responses. Impaired autophagy may also favor cell death by promoting apoptosis or necrosis. Acinar cell dedifferentiation permits proliferative, stem-like properties concomitant with loss of the acinar phenotype, and may give rise to tumorigenic intermediates. However, dedifferentiation must also be contemplated to achieve stem cell therapy, since adult tissues are non-proliferative.
5. References
- Acosta-Alvear D,Zhou Y,Blais A,Tsikitis M,Lents NH,Arias C, et al. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell 27(1): 53-66,2007. PMID: 17612490.
- Adachi Y,Yamamoto K,Okada T,Yoshida H,Harada A and Mori K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct Funct 33(1): 75-89,2008. PMID: 18360008.
- Alpy F,Rousseau A,Schwab Y,Legueux F,Stoll I,Wendling C, et al. STARD3 or STARD3NL and VAP form a novel molecular tether between late endosomes and the ER. J Cell Sci 126(Pt 23): 5500-5512,2013. PMID: 24105263.
- B'chir W,Maurin AC,Carraro V,Averous J,Jousse C,Muranishi Y, et al. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res,2013. PMID: 23804767.
- Beer S,Zhou J,Szabó A,Keiles S,Chandak GR,Witt H, et al. Comprehensive functional analysis of chymotrypsin C (CTRC) variants reveals distinct loss-of-function mechanisms associated with pancreatitis risk. Gut,2012. PMID: 22942235.
- Behnke J,Feige MJ and Hendershot LM. BiP and its nucleotide exchange factors Grp170 and Sil1: mechanisms of action and biological functions. J Mol Biol 427(7): 1589-1608,2015. PMID: 25698114.
- Berridge MJ. Inositol trisphosphate and calcium signalling. Nature 361(6410): 315-325,1993. PMID: 8381210.
- Bommiasamy H,Back SH,Fagone P,Lee K,Meshinchi S,Vink E, et al. ATF6alpha induces XBP1-independent expansion of the endoplasmic reticulum. J Cell Sci 122(Pt 10): 1626-1636,2009. PMID: 19420237.
- Brodsky JL and Wojcikiewicz RJ. Substrate-specific mediators of ER associated degradation (ERAD). Curr Opin Cell Biol 21(4): 516-521,2009. PMID: 19443192.
- Calfon M,Zeng H,Urano F,Till JH,Hubbard SR,Harding HP, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415(6867): 92-96,2002. PMID: 11780124.
- Collins MA,Yan W,Sebolt-Leopold JS and Pasca di Magliano M. MAPK signaling is required for dedifferentiation of acinar cells and development of pancreatic intraepithelial neoplasia in mice. Gastroenterology 146(3): 822-834 e827,2014. PMID: 24315826.
- Cox JS,Shamu CE and Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73(6): 1197-1206,1993. PMID: 8513503.
- Cross BC,Bond PJ,Sadowski PG,Jha BK,Zak J,Goodman JM, et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci U S A 109(15): E869-878,2012. PMID: 22315414.
- Cui H,Guo M,Xu D,Ding ZC,Zhou G,Ding HF, et al. The stress-responsive gene ATF3 regulates the histone acetyltransferase Tip60. Nat Commun 6: 6752,2015. PMID: 25865756.
- Eden ER,White IJ,Tsapara A and Futter CE. Membrane contacts between endosomes and ER provide sites for PTP1B-epidermal growth factor receptor interaction. Nat Cell Biol 12(3): 267-272,2010. PMID: 20118922.
- Fazio EN,Dimattia GE,Chadi SA,Kernohan KD and Pin CL. Stanniocalcin 2 alters PERK signalling and reduces cellular injury during cerulein induced pancreatitis in mice. BMC Cell Biol 12: 17,2011. PMID: 21545732.
- Fox RM and Andrew DJ. Transcriptional regulation of secretory capacity by bZip transcription factors. Front Biol (Beijing) 10(1): 28-51,2015. PMID: 25821458.
- Gerasimenko JV,Gerasimenko OV and Petersen OH. The role of Ca2+ in the pathophysiology of pancreatitis. J Physiol 592(Pt 2): 269-280,2014. PMID: 23897234.
- Goodall JC,Wu C,Zhang Y,McNeill L,Ellis L,Saudek V, et al. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc Natl Acad Sci U S A 107(41): 17698-17703,2010. PMID: 20876114.
- Guo L,Sans MD,Hou Y,Ernst SA and Williams JA. c-Jun/AP-1 is required for CCK-induced pancreatic acinar cell dedifferentiation and DNA synthesis in vitro. Am J Physiol Gastrointest Liver Physiol 302(12): G1381-1396,2012. PMID: 22461029.
- Hai T,Jalgaonkar S,Wolford CC and Yin X. Immunohistochemical detection of activating transcription factor 3, a hub of the cellular adaptive-response network. Methods Enzymol 490: 175-194,2011. PMID: 21266251.
- Harding HP,Novoa I,Zhang Y,Zeng H,Wek R,Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6(5): 1099-1108,2000. PMID: 11106749.
- Harding HP,Zhang Y and Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397(6716): 271-274,1999. PMID: 9930704.
- Haze K,Yoshida H,Yanagi H,Yura T and Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell 10(11): 3787-3799,1999. PMID: 10564271.
- Hollien J,Lin JH,Li H,Stevens N,Walter P and Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol 186(3): 323-331,2009. PMID: 19651891.
- Hollien J and Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313(5783): 104-107,2006. PMID: 16825573.
- Kowalik AS,Johnson CL,Chadi SA,Weston JY,Fazio EN and Pin CL. Mice lacking the transcription factor Mist1 exhibit an altered stress response and increased sensitivity to caerulein-induced pancreatitis. Am J Physiol Gastrointest Liver Physiol 292(4): G1123-1132,2007. PMID: 17170023.
- Kozutsumi Y,Segal M,Normington K,Gething MJ and Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332(6163): 462-464,1988. PMID: 3352747.
- Kubisch CH and Logsdon CD. Endoplasmic reticulum stress and the pancreatic acinar cell. Expert Rev Gastroenterol Hepatol 2(2): 249-260,2008. PMID: 19072360.
- Kubisch CH and Logsdon CD. Secretagogues differentially activate endoplasmic reticulum stress responses in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol 292(6): G1804-1812,2007. PMID: 17431218.
- Kubisch CH,Sans MD,Arumugam T,Ernst SA,Williams JA and Logsdon CD. Early activation of endoplasmic reticulum stress is associated with arginine-induced acute pancreatitis. Am J Physiol Gastrointest Liver Physiol 291(2): G238-245,2006. PMID: 16574987.
- Lee AH,Chu GC,Iwakoshi NN and Glimcher LH. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J 24(24): 4368-4380,2005. PMID: 16362047.
- Lee AH,Iwakoshi NN and Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23(21): 7448-7459,2003. PMID: 14559994.
- Lee AS,Bell J and Ting J. Biochemical characterization of the 94- and 78-kilodalton glucose-regulated proteins in hamster fibroblasts. J Biol Chem 259(7): 4616-4621,1984. PMID: 6707023.
- Li N,Wu X,Holzer RG,Lee JH,Todoric J,Park EJ, et al. Loss of acinar cell IKKα triggers spontaneous pancreatitis in mice. J Clin Invest 123(5): 2231-2243,2013. PMID: 23563314.
- Lu M,Lawrence DA,Marsters S,Acosta-Alvear D,Kimmig P,Mendez AS, et al. Cell death. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 345(6192): 98-101,2014. PMID: 24994655.
- Lugea A,Tischler D,Nguyen J,Gong J,Gukovsky I,French SW, et al. Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology 140(3): 987-997,2011. PMID: 21111739.
- Lugea A,Waldron RT,French SW and Pandol SJ. Drinking and driving pancreatitis: links between endoplasmic reticulum stress and autophagy. Autophagy 7(7): 783-785,2011. PMID: 21460613.
- Luo X,Shin DM,Wang X,Konieczny SF and Muallem S. Aberrant localization of intracellular organelles, Ca2+ signaling, and exocytosis in Mist1 null mice. J Biol Chem 280(13): 12668-12675,2005. PMID: 15665001.
- Malhi H,Kropp EM,Clavo VF,Kobrossi CR,Han J,Mauer AS, et al. C/EBP Homologous Protein-induced Macrophage Apoptosis Protects Mice from Steatohepatitis. J Biol Chem 288(26): 18624-18642,2013. PMID: 23720735.
- Malo A,Krüger B,Göke B and Kubisch CH. 4-Phenylbutyric acid reduces endoplasmic reticulum stress, trypsin activation, and acinar cell apoptosis while increasing secretion in rat pancreatic acini. Pancreas 42(1): 92-101,2013. PMID: 22889983.
- Malo A,Krüger B,Seyhun E,Schäfer C,Hoffmann RT,Göke B, et al. Tauroursodeoxycholic acid reduces endoplasmic reticulum stress, trypsin activation, and acinar cell apoptosis while increasing secretion in rat pancreatic acini. Am J Physiol Gastrointest Liver Physiol 299(4): G877-886,2010. PMID: 20671193.
- Marciniak SJ,Yun CY,Oyadomari S,Novoa I,Zhang Y,Jungreis R, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 18(24): 3066-3077,2004. PMID: 15601821.
- Maurel M,Chevet E,Tavernier J and Gerlo S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem Sci 39(5): 245-254,2014. PMID: 24657016.
- McCullough KD,Martindale JL,Klotz LO,Aw TY and Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 21(4): 1249-1259,2001. PMID: 11158311.
- Medina DL,Di Paola S,Peluso I,Armani A,De Stefani D,Venditti R, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 17(3): 288-299,2015. PMID: 25720963.
- Mori K,Ma W,Gething MJ and Sambrook J. A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 74(4): 743-756,1993. PMID: 8358794.
- Nikawa J and Yamashita S. IRE1 encodes a putative protein kinase containing a membrane-spanning domain and is required for inositol phototrophy in Saccharomyces cerevisiae. Mol Microbiol 6(11): 1441-1446,1992. PMID: 1625574.
- Oyadomari S and Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 11(4): 381-389,2004. PMID: 14685163.
- Pandol SJ,Gorelick FS and Lugea A. Environmental and genetic stressors and the unfolded protein response in exocrine pancreatic function - a hypothesis. Front Physiol 2: 8,2011. PMID: 21483727.
- Patel S and Muallem S. Acidic Ca(2+) stores come to the fore. Cell Calcium 50(2): 109-112,2011. PMID: 21497395.
- Penny CJ,Kilpatrick BS,Eden ER and Patel S. Coupling acidic organelles with the ER through Ca(2+) microdomains at membrane contact sites. Cell Calcium 58(4): 387-396,2015. PMID: 25866010.
- Pin CL,Rukstalis JM,Johnson C and Konieczny SF. The bHLH transcription factor Mist1 is required to maintain exocrine pancreas cell organization and acinar cell identity. J Cell Biol 155(4): 519-530,2001. PMID: 11696558.
- Puthalakath H,O'Reilly LA,Gunn P,Lee L,Kelly PN,Huntington ND, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129(7): 1337-1349,2007. PMID: 17604722.
- Rocha N,Kuijl C,van der Kant R,Janssen L,Houben D,Janssen H, et al. Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 Glued and late endosome positioning. J Cell Biol 185(7): 1209-1225,2009. PMID: 19564404.
- Ronco V,Potenza DM,Denti F,Vullo S,Gagliano G,Tognolina M, et al. A novel Ca(2)(+)-mediated cross-talk between endoplasmic reticulum and acidic organelles: implications for NAADP-dependent Ca(2)(+) signalling. Cell Calcium 57(2): 89-100,2015. PMID: 25655285.
- Rouschop KM,van den Beucken T,Dubois L,Niessen H,Bussink J,Savelkouls K, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest 120(1): 127-141,2010. PMID: 20038797.
- Sah RP,Dawra RK and Saluja AK. New insights into the pathogenesis of pancreatitis. Curr Opin Gastroenterol 29(5): 523-530,2013. PMID: 23892538.
- Sah RP,Garg SK,Dixit AK,Dudeja V,Dawra RK and Saluja AK. Endoplasmic reticulum stress is chronically activated in chronic pancreatitis. J Biol Chem 289(40): 27551-27561,2014. PMID: 25077966.
- Schindler AJ and Schekman R. In vitro reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc Natl Acad Sci U S A 106(42): 17775-17780,2009. PMID: 19822759.
- Seyhun E,Malo A,Schäfer C,Moskaluk CA,Hoffmann RT,Göke B, et al. Tauroursodeoxycholic acid reduces endoplasmic reticulum stress, acinar cell damage, and systemic inflammation in acute pancreatitis. Am J Physiol Gastrointest Liver Physiol 301(5): G773-782,2011. PMID: 21778463.
- Shaffer AL,Shapiro-Shelef M,Iwakoshi NN,Lee AH,Qian SB,Zhao H, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 21(1): 81-93,2004. PMID: 15345222.
- Shi G,DiRenzo D,Qu C,Barney D,Miley D and Konieczny SF. Maintenance of acinar cell organization is critical to preventing Kras-induced acinar-ductal metaplasia. Oncogene 32(15): 1950-1958,2013. PMID: 22665051.
- Shoulders MD,Ryno LM,Genereux JC,Moresco JJ,Tu PG,Wu C, et al. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep 3(4): 1279-1292,2013. PMID: 23583182.
- Sidrauski C and Walter P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90(6): 1031-1039,1997. PMID: 9323131.
- Streb H,Irvine RF,Berridge MJ and Schulz I. Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature 306(5938): 67-69,1983. PMID: 6605482.
- Suyama K,Ohmuraya M,Hirota M,Ozaki N,Ida S,Endo M, et al. C/EBP homologous protein is crucial for the acceleration of experimental pancreatitis. Biochem Biophys Res Commun 367(1): 176-182,2008. PMID: 18166146.
- Tabas I and Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 13(3): 184-190,2011. PMID: 21364565.
- Upton JP,Wang L,Han D,Wang ES,Huskey NE,Lim L, et al. IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 338(6108): 818-822,2012. PMID: 23042294.
- Urano F,Wang X,Bertolotti A,Zhang Y,Chung P,Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287(5453): 664-666,2000. PMID: 10650002.
- Venkatachalam K,Wong CO and Montell C. Feast or famine: role of TRPML in preventing cellular amino acid starvation. Autophagy 9(1): 98-100,2013. PMID: 23047439.
- Vidal RL and Hetz C. Unspliced XBP1 controls autophagy through FoxO1. Cell Res 23(4): 463-464,2013. PMID: 23337584.
- Wang S and Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol 197(7): 857-867,2012. PMID: 22733998.
- Wang WA,Groenendyk J and Michalak M. Calreticulin signaling in health and disease. Int J Biochem Cell Biol 44(6): 842-846,2012. PMID: 22373697.
- Wu L,Cai B,Zheng S,Liu X,Cai H and Li H. Effect of Emodin on Endoplasmic Reticulum Stress in Rats with Severe Acute Pancreatitis. Inflammation,2013. PMID: 23605470.
- Ye R,Mareninova OA,Barron E,Wang M,Hinton DR,Pandol SJ, et al. Grp78 heterozygosity regulates chaperone balance in exocrine pancreas with differential response to cerulein-induced acute pancreatitis. Am J Pathol 177(6): 2827-2836,2010. PMID: 20971738.
- Yoshida H,Haze K,Yanagi H,Yura T and Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem 273(50): 33741-33749,1998. PMID: 9837962.
- Yoshida H,Matsui T,Yamamoto A,Okada T and Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107(7): 881-891,2001. PMID: 11779464.
- Yoshida H,Oku M,Suzuki M and Mori K. pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J Cell Biol 172(4): 565-575,2006. PMID: 16461360.
- Yoshida Y and Tanaka K. Lectin-like ERAD players in ER and cytosol. Biochim Biophys Acta 1800(2): 172-180,2010. PMID: 19665047.
- Zeng Y,Wang X,Zhang W,Wu K and Ma J. Hypertriglyceridemia aggravates ER stress and pathogenesis of acute pancreatitis. Hepatogastroenterology 59(119): 2318-2326,2012. PMID: 22389298.