
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2016.5
| Attachment | Size |
|---|---|
| 422.06 KB | |
| 162.63 KB |
Bile acids (BAs) are natural end products of cholesterol metabolism (12). The physiological functions of BAs are the emulsification of lipid aggregates and solubilization of lipids in an aqueous environment. The major BAs in humans are chenodeoxycholic acid (CDCA) and cholic acid (CA), which are known as primary BAs since they are synthesized in the liver (46). Before secretion by hepatocytes, primary BAs are conjugated with either taurine or glycine, which increases their polarity and water solubility. Secondary bile acids such as deoxycholic acid (DCA) and lithocholic acid (LCA) are produced in the colon by bacterial dehydroxylation of the primary BAs. Under physiological conditions, BAs are temporarily stored in the gallbladder and are released to the intestine. Most of the BAs are then efficiently reabsorbed from the ileum and transported back to the liver via the portal vein (enterohepatic circulation). Under normal, physiological conditions, BAs cannot get into the pancreas. However, under pathophysiological conditions, such as obstruction of the ampulla of Vater by an impacted gallstone, bile can enter into the pancreatic ducts and trigger pancreatitis (32). Unfortunately, we do not know the concentration of bile acids that can reach the pancreatic ductal cells under pathological conditions. It probably varies among patients and mainly depends on the duration of ampullary gallstone obstruction. However, previous studies have shown that relatively low concentrations of BAs (25-200 µM) are able to cause intracellular Ca2+ signalling and cell death in acinar cells (23, 49). The close relationship between the passage of a gallstone and the development of acute pancreatitis (AP) has been known for more than a hundred years (32) and has been confirmed in a number of studies (30, 33, 45). However; the pathogenesis underlying the development of biliary AP is not well understood.
Most of the research investigating the pathomechanism of AP has been done on pancreatic acinar cells. These studies have demonstrated that the central intra-acinar events in pancreatitis are elevation of intra-acinar Ca2+ concentration, premature activation of trypsinogen and the activation of the proinflammatory transcription factor, nuclear factor-κB (NF-κB) which leads to pancreatic injury (6, 16-18, 36, 44). It has been shown that one of the most toxic BAs to acinar cells is the secondary BA, taurolithocholic acid (TLC) that forms from LCA after re-absorption from the intestine. The sulfated form of TLC (TLCS) causes Ca2+ signalling in pancreatic acinar cells via an inositol 1,4,5-trisphosphate (IP3)-dependent mobilization of sequestered intracellular Ca2+ (49). The elevated [Ca2+]i can lead to enzyme activation (36) and/or cell death (23) and result in severe acute necrotizing pancreatitis. Since increased [Ca2+] level is a critical step in the initiation of acinar cell injury, several studies have focused on the prevention of cytosolic calcium overload. The calcium chelator, caffeine and its dimethylxanthine metabolites, efficiently decreased TLCS-induced calcium elevation and cell death on isolated pancreatic acinar cells. Caffeine also reduced the severity of TLCS-induced AP, most probably by the inhibition of IP3R-mediated calcium signalling (20). In another aspect, blocking of Ca2+ entry by pharmacological inhibition of store-operated Ca2+ channel, Orai-1 has also proven to be effective against the TLCS-induced acinar cell necrosis (52).The target of the sustained Ca2+ signal is the Ca2+-activated phosphatase, calcineurin, which plays a central role in the intra-acinar activation of zymogens and NF-κB (27). In addition to Ca2+, other cellular mechanisms are also involved in the BA-induced acinar injury, such as mitochondrial dysfunction (28, 50), depletion of cytosolic ATP(51) or increased reactive oxygen species (ROS) production (7).
In contrast to acinar cells, the role of pancreatic ductal epithelial cells (PDECs) in the pathogenesis of biliary AP has received much less attention, despite being the first pancreatic cell type exposed to the refluxed bile. Pancreatic ducts can be divided into three main types on the basis of their size and location. These are the main duct, interlobular and intralobular ducts. The main duct mostly collects and drains the juice secreted by other branches of the ductal tree, whereas intra/interlobular ducts are the main sites of HCO3- secretion. Although, several studies have shown that ductal fluid and HCO3- secretion are crucially important to maintain the integrity of the pancreas (4, 10, 11, 19), the role of PDECs in the development of AP has only been highlighted recently. In vivo studies have shown that pancreatic hypersecretion (with hypoproteinaemia) occurs in the early phase of AP, which develops into hyposecretion during the onset of pancreatitis (10, 11, 19). This hypersecretion may represent a defence mechanism by washing out the toxic factors from the pancreas. The beneficial effect of fluid hypersecretion is further supported by studies in which secretin, one of the major secretagogues of ductal fluid secretion, was shown to reduce the severity of caerulein-induced AP (40, 41). In addition, impaired or insufficient ductal fluid secretion, such as observed in cystic fibrosis, increases the risk of AP (13, 14). Taken together, these data strongly suggest that PDECs represent an important and essential protective mechanism in the exocrine pancreas.
1. Effect of Bile Acids on the Main Pancreatic Duct
In the 1980s, it was postulated that the breakdown of the pancreatic duct permeability barrier is a risk factor for the development of AP. Therefore, researchers extensively investigated the effect of BAs on the morphology and permeability of the main pancreatic duct. In these in vivo studies various BAs were perfused through the cannulated main duct and the permeability of the pancreatic duct mucosal barrier was measured using different techniques (5, 15, 37-39). It has been shown that BAs in high concentrations (2-15 mM), made the ducts permeable to molecules as large as 20,000-daltons, whereas they are normally impermeable to molecules over 3,000-daltons (5, 15). BAs in mM concentrations also increase the permeability of the main duct to Cl- and HCO3- (37-39). The effect of dihydroxy BAs was significantly greater than the effect of trihydroxy BAs on the permeability of these anions, most probably because trihydroxy BAs are less lipid soluble and therefore less toxic to the cells. The changes in ductal permeability were in accord with the changes in the morphology of the ductal epithelia. Perfusion with higher concentrations of BAs (15 mM) caused disruption of cell integrity, flattening of the ductal epithelium and cell loss (5).Due to their detergent properties, this harmful effect of BAs is not surprising.
It was also highlighted that infected bile is more harmful to the duct cells than sterile bile (5, 37, 38). The higher toxicity of infected bile is probably due to bacterial deconjugation, which produces more toxic unconjugated BAs. The toxicity of BAs mainly depends on their solubility and the degree of ionisation. At neutral pH, unconjugated bile acids exist in an unionized (9), electrically neutral, form and therefore can pass easily through the cell membrane. In contrast, glycine- and taurine-conjugated BAs, which have a lower pKa (around 4 and 2 respectively), are ionized at neutral pH (9) and are therefore less lipid soluble.
Although, in vivo animal studies have a very important significance, their relevance to human disease is doubtful. One of the major problems with these studies is that BAs were used in relatively high concentrations; probably higher than would be present in the pancreatic duct if reflux occurs. Moreover, at these extremely high concentrations, BAs caused excessive and uncontrolled destruction of both acini and ducts.
In vitro studies have allowed the investigation of more pathophysiologically relevant effects of BAs on the ductal epithelium. Okolo et al. studied the effects of BAs (100 µM – 2 mM) on the ion conductances and monolayer resistance of cultured PDECs isolated from the accessory pancreatic duct of dog (31). They found that taurodeoxycholic acid (TDCA) and taurochenodeoxycholic acid caused a concentration-dependent increase in both Cl- and K+ conductances, whereas the trihydroxy BA, taurocholic acid, was completely ineffective. The increases in Cl- and K+ conductances were mediated via elevation of [Ca2+]i and blocked by 4,4,-diisothiocyanostilbene-2,2,-disulfonic acid (DIDS) and charybdotoxin, respectively. Using Ussing chambers, they could localize the Cl- conductance to the apical, and the K+ conductance to the basolateral membrane of PDECs. In addition, they showed that only higher concentrations of BAs decreased the monolayer transepithelial resistance. Similar results have been found in bovine PDECs, where TDC markedly increased transepithelial ion transport and decreased the electrical resistance of the tissue (1). On the other hand, TDC caused dose-dependent mucosal damage (2) and at higher concentrations extensive loss of the epithelial cell lining (1).
2. Effect of Bile Acids on the Intra/Interlobular Pancreatic Ducts
Although, the earlier studies described above, characterized the effects of BAs on the permeability and morphology of the main pancreatic duct, no information was available about their effects on the smaller ducts. However, the development of microdissection techniques for the isolation of small intra/interlobular ducts (3), led to a break-through in our understanding of the cellular physiology of the ductal cells.
The major physiological function of the intra/interlobular pancreatic ductal cells is to secrete a HCO3--rich alkaline fluid that washes digestive enzymes out of the gland and neutralizes acid chyme in the duodenum (3). The effects of BAs on HCO3--secretion have been intensively investigated in the last few years (26, 47, 48), and these studies suggest that the role of ductal cells in the pathomechanism of biliary AP is complex.
Our research group has shown that both basolateral and luminal administration of either non-conjugated or glycine-conjugated forms of CDCA causes a dose-dependent intracellular acidification in guinea pig PDECs (48). Interestingly, basolateral administration of 1 mM CDCA for 6–8 min damaged the membrane integrity, and the cells lost the fluorescent dye, very quickly. The same concentration of CDCA on the luminal membrane had no toxic effects. Okolo et al. also found differences between the effects of BAs on the luminal and basolateral membranes (31). In addition, both CDCA and glycochenodeoxycholate (GCDCA) induced a dose-dependent increase in [Ca2+]i via phospholipase C- and IP3 receptor-mediated mechanisms. GCDCA had a smaller effect on intracellular pH (pHi) and (Ca2+)i than CDCA, most probably because conjugated BAs are ionised at neutral pH and therefore require active transport mechanisms for cellular uptake.
We also found that the effect of CDCA on ductal HCO3- efflux depends on its concentration (48). At low concentration (0.1 mM), CDCA significantly stimulated HCO3- efflux by a DIDS–sensitive Cl-/HCO3- exchange mechanism. The stimulatory effect of CDCA was observed only when CDCA was added to the lumen of the ducts and was dependent on Ca2+mobilization. In contrast, high concentration of CDCA (1 mM) caused pathological Ca2+ signalling and strongly inhibited HCO3- efflux. This inhibitory effect of high concentration of CDCA was independent of changes in [Ca2+]i and was observed when CDCA was applied to either the luminal or the basolateral membrane of the ducts (48). The effect of the conjugated GCDCA on pHi and [Ca2+]i suggest that although GCDCA can enter the cells, most probably by a transporter-mediated mechanism, it had no effect on HCO3- efflux at both high and low concentrations.
The differences in the effects of low and high concentrations of CDCA suggest that non-conjugated BAs have a specific mode of action on PDECs which strongly depends on their concentration. The key question is the identification of the cellular mechanisms by which BAs exert these opposite effects. Perides et al. have recently identified the presence of a G-protein-coupled bile acid receptor-1 (GPBAR1also known as TGR5) on the apical membrane of acinar cells (34). They showed that GPBAR 1 knockout mice were completely protected against TLCS-induced pancreatitis and suggested that this receptor has a central role in the BA-induced acinar cell injury in mice. In contrast, guinea pig pancreatic ductal cells do not express GPBAR1 (47) suggesting that this receptor is not involved in the effect of CDCA on PDECs.
A) Stimulatory effect of low concentrations of bile acids
Since CDCA only increased HCO3- efflux when applied to the luminal membrane, it is likely that the stimulatory effect of CDCA is due to activation of one, or more, Ca2+-dependent apical transporters. The SLC26 anion transporters (29, 53) and the Ca2+-activated Cl- channel (CaCC) are known to be activated by an increase in [Ca2+]i suggesting that these transporters could be the target for the CDCA-induced increase in Ca2+.
Using the whole cell configuration of the patch clamp technique, CDCA failed to activate CaCC, but induced a robust and reversible increase in K+ currents (47). The activated currents could be blocked by the specific large-conductance Ca2+-activated K+ channel (BK) inhibitor, iberiotoxin. In contrast, the small- and intermediate Ca2+-activated K+ channel inhibitors, UCL1684 and TRAM34 had no effect on the CDCA-activated currents. Luminal administration of iberiotoxin completely blocked the stimulatory effect of CDCA on HCO3- efflux in microperfused ducts. In contrast, basolateral iberiotoxin had no effect on the luminal CDCA-stimulated HCO3- efflux. These data strongly indicate that BK channels play a central role in the stimulatory effect of CDCA on HCO3- efflux and that they are localized to the luminal membrane of the ductal cells. This latter hypothesis has been confirmed by immunohistochemistry which showed strong expression of BK channels at the apical membrane of guinea pig intra/interlobular ducts (47). Moreover, activation of BK channels by luminal administration of a pharmacological compound, NS11021, increased HCO3- efflux in a similar manner to CDCA. Our hypothesis is that activation of apical BK channels leads to the hyperpolarisation of the apical plasma membrane, which in turn increases the electrochemical driving force for anion efflux through SLC26 anion exchangers (Figure 1). The CFTR Cl- channel is one of the major ion channels on the apical membrane of PDECs, which interacts with the Cl-/ HCO3- exchanger, therefore plays an essential role in HCO3- efflux. The possible role of this ion channel has also been raised in the stimulatory effect of CDCA (21). CFPAC-1 is a human, pancreatic ductal cell line which deficient for CFTR, but expresses the SLC26A6 anion exchanger. Administration of 0.1 mM CDCA had no effect on HCO3- efflux in these cells.
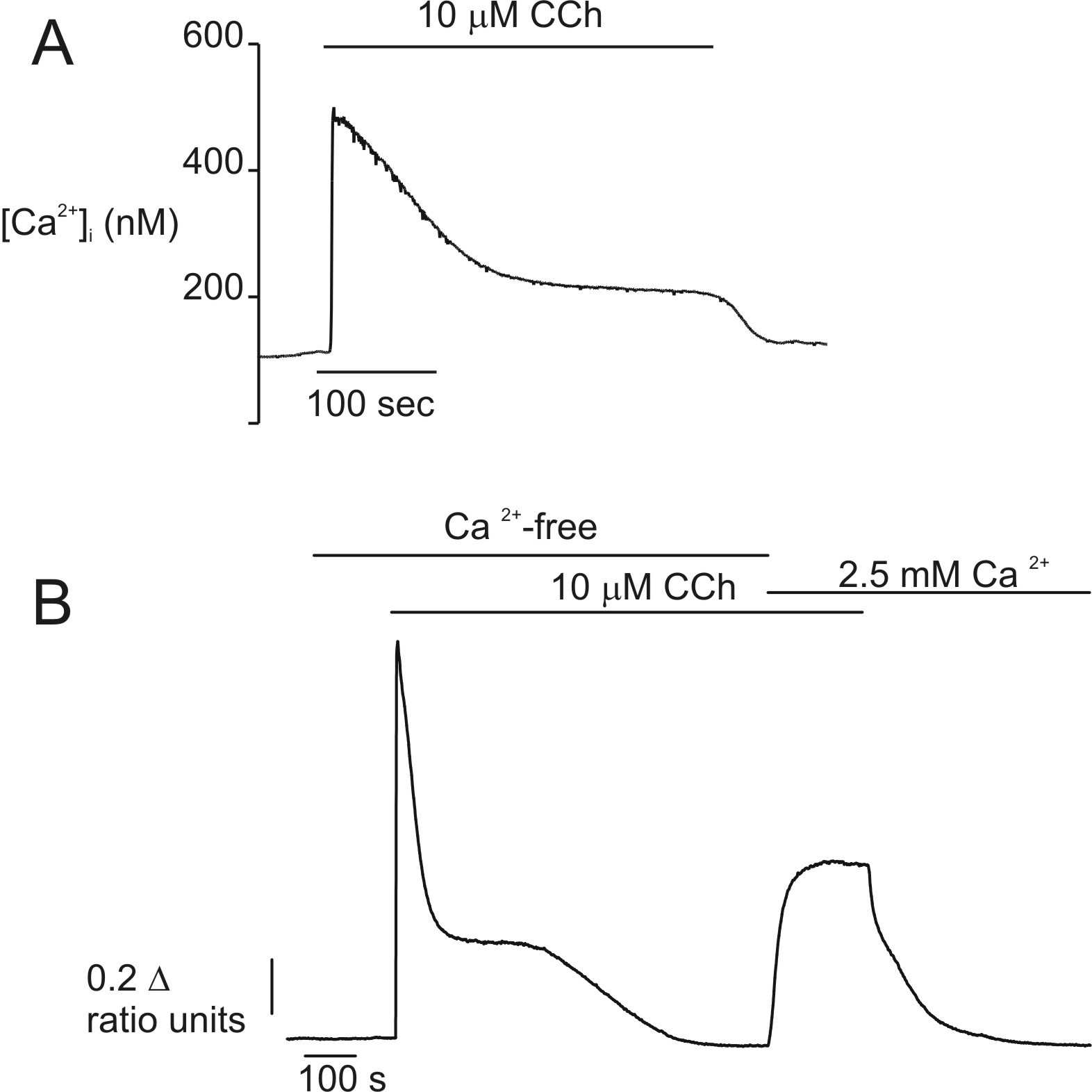
Figure 1. Cellular mechanism of the stimulatory effect of CDCA. Low concentration of CDCA (0.1 mM) induces an elevation of (Ca2+)i by two distinct mechanisms: (1) release of (Ca2+)i from the endoplasmic reticulum by an IP3R- and PLC-mediated pathway and (2) extracellular ATP-induced Ca2+ influx directly and indirectly through P2X and P2Y receptors, respectively. The increase in [Ca2+]i activates BK channels that leads to the hyperpolarisation of the plasma membrane which in turn increases the electrochemical driving force for anion secretion through CFTR and SLC26 anion exchangers. BK: large conductance Ca2+-activated K+ channel, Ca2+: calcium, CDCA: chenodeoxycholic acid, CFTR: cystic fibrosis transmembrane conductance regulator Cl- channel, Cx: connexin, ER: endoplasmic reticulum, IP3R: inositol 1,4,5-trisphosphate receptor, Px: pannexin. +: stimulation.
However; after the transduction of CFPAC-1 cells with wild-type CFTR, CDCA significantly stimulated HCO3- efflux. CDCA had no effect on the activity of this channel, as demonstrated by the patch clamp technique, although it requires CFTR expression to exert its stimulatory effect (21).
A recent study by Kowal et al. has indicate that in the stimulatory effect of CDCA, release of ATP and activation of purinergic receptors are also involved (24, 25). Using a pancreatic ductal cell line (Capan-1) they showed that CDCA induces a dose-dependent release of ATP which includes both vesicular and non-vesicular mechanism and is more pronounced when CDCA was applied from the luminal membrane. Extracellular ATP than binds to P2 receptors leading to increases in [Ca2+]i which may be involved in the activation of Ca2+-activated ion channels, such as BK channels (Figure 1). It has been also shown that Capan-1 cells express GPBAR1 receptor which is activated by CDCA; although activation of this receptor is not involved in the CDCA-induced ATP release.
The stimulatory effect of low concentrations of CDCA highlights the importance of ductal fluid secretion in the protection of the pancreas. The increased volume of fluid can be beneficial in several ways:
(i) High concentrations of HCO3- in the secreted fluid promote the deprotonation of BAs to less toxic bile salts.
(ii) The increased volume of fluid decreases the concentration of BAs in the ducts.
(iii) The greater ductal flow may push stones through the papilla of Vater to clear the obstruction.
(iv) Increased fluid secretion may wash out the toxic BAs from the ductal tree in order to avoid pancreatic injury.
B) Inhibitory effect of high concentrations of bile acids
If the stimulated secretion is not able to wash out BAs from the ductal tree, the luminal concentrations of BAs will increase further. In this situation, high concentrations of CDCA cause pathologic Ca2+ signalling and inhibition of the acid/base transporters of PDECs (48). We have provided evidence that mitochondrial damage and depletion of intracellular ATP (ATPi) are the most crucial factors in the toxic effect of CDCA on pancreatic ductal secretion (26). Administration of 1 mM CDCA to PDECs for 10 minutes caused swelling of the mitochondria and disruption of their inner membranes. Damage of the mitochondria markedly and irreversibly reduced ATPi. Exposure of pancreatic ducts to carbonyl cyanide m-chlorophenyl hydrazone and deoxyglucose/iodoacetamide (inhibitors of oxidative phosphorylation and the glycolytic pathway respectively) totally mimicked the effect of 1 mM CDCA. These data indicate that CDCA inhibits both the oxidative and glycolytic pathways in PDECs. In addition, it has been shown that ATPi depletion is crucial in the inhibitory effect of CDCA on ductal ion transport mechanisms. In the absence of ATPi the acid/base transporters do not work properly, which leads to impaired fluid secretion and finally cell death. (Figure 2) In this case, BAs could reach the acinar cells, either by diffusion up the ductal tree or by leakage into the gland interstitium, where they will switch on pathologic Ca2+ signalling and trigger AP. We have also shown that the CDCA-induced mitochondrial injury is due to the opening of mitochondrial permeability transition pore (mPTP), which leads to mitochondrial swelling/dysfunction and consequently inhibition of ATP synthesis (22). Studies on hepatocytes have demonstrated that the toxic effect of hydrophobic BAs can be attenuated by the hydrophilic BA, ursodeoxycholic acid (UDCA) (8, 35, 42, 43). These studies have shown that the cytoprotective effect of UDCA is largely based on its ability to stabilize the mitochondrial membrane by the inhibition of BA-induced mPTP opening. 24 h pre-treatment of the pancreatic ducts with 0.5 mM UDCA significantly decreased the CDCA-induced mitochondrial injury by the inhibition of CDCA-induced mPTP and ATPi loss and reduced the inhibitory effect of CDCA on the acid/base transporters (22). Moreover, the protective effect of UDCA on the mitochondria is associated with anti-apoptotic effect which raises the possible therapeutic use of UDCA in the treatment of bile-induced AP.
3. Conclusion
Taken together, both in vivo and in vitro studies indicate that once BAs reach the ductal epithelium, depending on their concentration, they exert a harmful or beneficial effect on PDECs (Table 1). This biphasic effect of BAs on ductal secretion may be a significant factor in the pathomechanism of biliary AP.

Figure 2. Cellular mechanism of the inhibitory effect of CDCA. High concentration of CDCA (1 mM) induces toxic (Ca2+)i signal, mitochondrial damage and depletion of ATPi that leads to the inhibition of acid-base transporters of PDEC. 24 h pre-treatment of the ducts with UDCA (0.5 mM) is able to prevent the toxic effect of CDCA by the stabilization of the mitochondrial membrane. BK: large conductance Ca2+-activated K+ channel, Ca2+: calcium, CDCA: chenodeoxycholic acid, CFTR: cystic fibrosis transmembrane conductance regulator Cl- channel, Cx: connexin, ER: endoplasmic reticulum, IP3R: inositol 1,4,5-trisphosphate receptor, Px: pannexin, UDCA: ursodeoxycholic acid. -: inhibition.

Table 1. Summary of the effect of bile acids on pancreatic ductal epithelial cells. CA: cholic acid, CDCA: chenodeoxycholic acid, DCA: deoxycholic acid, GCA: glycocholic acid, GDCA: glycodeoxycholic acid, GCDCA: glycochenodeoxycholic acid, TCA: taurocholic acid, TCDCA: taurochenodeoxycholic acid, TDCA: taurodeoxycholic acid.
4. References
- Alvarez C,Fasano A and Bass BL. Acute effects of bile acids on the pancreatic duct epithelium in vitro. J Surg Res 74(1): 43-46,1998. PMID 9536972.
- Alvarez C,Nelms C,D'Addio V and Bass BL. The pancreatic duct epithelium in vitro: bile acid injury and the effect of epidermal growth factor. Surgery 122(2): 476-483; discussion 483-474,1997. PMID 9288155.
- Argent BE,Arkle S,Cullen MJ and Green R. Morphological, biochemical and secretory studies on rat pancreatic ducts maintained in tissue culture. Q J Exp Physiol 71(4): 633-648,1986. PMID: 3024200.
- Argent BE, Gray MA, Steward MC, Case RM. Cell Physiology of Pancreatic Ducts. Physiology of the Gastrointestinal Tract. LR Johnson. San Diego, Elsevier. 2:1376-1396, 2006.
- Armstrong CP,Taylor TV and Torrance HB. Effects of bile, infection and pressure on pancreatic duct integrity. Br J Surg 72(10): 792-795,1985. PMID: 3899241.
- Bialek R,Willemer S,Arnold R and Adler G. Evidence of intracellular activation of serine proteases in acute cerulein-induced pancreatitis in rats. Scand J Gastroenterol 26(2): 190-196,1991. PMID: 1707179.
- Booth DM,Murphy JA,Mukherjee R,Awais M,Neoptolemos JP,Gerasimenko OV, et al. Reactive oxygen species induced by bile acid induce apoptosis and protect against necrosis in pancreatic acinar cells. Gastroenterology 140(7): 2116-2125,2011. PMID: 21354148.
- Botla R,Spivey JR,Aguilar H,Bronk SF and Gores GJ. Ursodeoxycholate (UDCA) inhibits the mitochondrial membrane permeability transition induced by glycochenodeoxycholate: a mechanism of UDCA cytoprotection. J Pharmacol Exp Ther 272(2): 930-938,1995. PMID: 7853211.
- Carey J. Bile salt metabolism in man. The Bile Acids. Chemistry, Physiology, and Metabolism. P Nair, Kritchevsky D. New York, Plenum Pub Corp. 2:55, 1973.
- Czako L,Yamamoto M and Otsuki M. Exocrine pancreatic function in rats after acute pancreatitis. Pancreas 15(1): 83-90,1997. PMID: 9211497.
- Czako L,Yamamoto M and Otsuki M. Pancreatic fluid hypersecretion in rats after acute pancreatitis. Dig Dis Sci 42(2): 265-272,1997. PMID: 9052504.
- Danielsson H and Sjovall J. Bile acid metabolism. Annu Rev Biochem 44: 233-253,1975. PMID: 1094911.
- Durie PR. Pancreatitis and mutations of the cystic fibrosis gene. N Engl J Med 339(10): 687-688,1998. PMID: 9725928.
- Durie PR. The pathophysiology of the pancreatic defect in cystic fibrosis. Acta Paediatr Scand Suppl 363: 41-44,1989. PMID: 2701923.
- Farmer RC,Tweedie J,Maslin S,Reber HA,Adler G and Kern H. Effects of bile salts on permeability and morphology of main pancreatic duct in cats. Dig Dis Sci 29(8): 740-751,1984. PMID: 6745035.
- Grady T,Mah'Moud M,Otani T,Rhee S,Lerch MM and Gorelick FS. Zymogen proteolysis within the pancreatic acinar cell is associated with cellular injury. Am J Physiol Gastrointest Liver Physiol 275(5 Pt 1): G1010-1017,1998. PMID: 9815031.
- Grady T,Saluja A,Kaiser A and Steer M. Edema and intrapancreatic trypsinogen activation precede glutathione depletion during caerulein pancreatitis. Am J Physiol Gastrointest Liver Physiol 271(1 Pt 1): G20-26,1996. PMID: 8760102.
- Gukovsky I,Gukovskaya AS,Blinman TA,Zaninovic V and Pandol SJ. Early NF-kappaB activation is associated with hormone-induced pancreatitis. Am J Physiol Gastrointest Liver Physiol 275(6 Pt 1): G1402-1414,1998. PMID: 9843778.
- Hegyi P,Czako L,Takacs T,Szilvassy Z and Lonovics J. Pancreatic secretory responses in L-arginine-induced pancreatitis: comparison of diabetic and nondiabetic rats. Pancreas 19(2): 167-174,1999. PMID: 10438164.
- Huang W,Cane MC,Mukherjee R,Szatmary P,Zhang X,Elliott V, et al. Caffeine protects against experimental acute pancreatitis by inhibition of inositol 1,4,5-trisphosphate receptor-mediated Ca2+ release. Gut,2015. PMID: 26642860.
- Ignath I,Hegyi P,Venglovecz V,Szekely CA,Carr G,Hasegawa M, et al. CFTR expression but not Cl- transport is involved in the stimulatory effect of bile acids on apical Cl-/HCO3- exchange activity in human pancreatic duct cells. Pancreas 38(8): 921-929,2009. PMID: 19752774.
- Katona M,Hegyi P,Kui B,Balla Z,Rakonczay Z, Jr.,Razga Z, et al. A novel, protective role of ursodeoxycholate in bile-induced pancreatic ductal injury. Am J Physiol Gastrointest Liver Physiol 310(3): G193-204,2016. PMID: 26608189.
- Kim JY,Kim KH,Lee JA,Namkung W,Sun AQ,Ananthanarayanan M, et al. Transporter-mediated bile acid uptake causes Ca2+-dependent cell death in rat pancreatic acinar cells. Gastroenterology 122(7): 1941-1953,2002. PMID: 12055600.
- Kowal JM,Haanes KA,Christensen NM and Novak I. Bile acid effects are mediated by ATP release and purinergic signalling in exocrine pancreatic cells. Cell Commun Signal 13: 28,2015. PMID: 26050734.
- Kowal JM,Yegutkin GG and Novak I. ATP release, generation and hydrolysis in exocrine pancreatic duct cells. Purinergic Signal 11(4): 533-550,2015. PMID: 26431833.
- Maleth J,Venglovecz V,Razga Z,Tiszlavicz L,Rakonczay Z, Jr. and Hegyi P. Non-conjugated chenodeoxycholate induces severe mitochondrial damage and inhibits bicarbonate transport in pancreatic duct cells. Gut 60(1): 136-138,2011. PMID: 20732916.
- Muili KA,Jin S,Orabi AI,Eisses JF,Javed TA,Le T, et al. Pancreatic acinar cell nuclear factor kappaB activation because of bile acid exposure is dependent on calcineurin. J Biol Chem 288(29): 21065-21073,2013. PMID: 23744075.
- Mukherjee R,Mareninova OA,Odinokova IV,Huang W,Murphy J,Chvanov M, et al. Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: inhibition prevents acute pancreatitis by protecting production of ATP. Gut,2015. PMID: 26071131.
- Namkung W,Lee JA,Ahn W,Han W,Kwon SW,Ahn DS, et al. Ca2+ activates cystic fibrosis transmembrane conductance regulator- and Cl- -dependent HCO3 transport in pancreatic duct cells. J Biol Chem 278(1): 200-207,2003. PMID: 12409301.
- Niederau C,Niederau M,Luthen R,Strohmeyer G,Ferrell LD and Grendell JH. Pancreatic exocrine secretion in acute experimental pancreatitis. Gastroenterology 99(4): 1120-1127,1990. PMID: 2394333.
- Okolo C,Wong T,Moody MW and Nguyen TD. Effects of bile acids on dog pancreatic duct epithelial cell secretion and monolayer resistance. Am J Physiol Gastrointest Liver Physiol 283(5): G1042-1050,2002. PMID: 12381517.
- Opie E. The etiology of acute haemorrhagic pancreatitis. Johns Hopkins Hospital Bulletin (12): 182-188,1901.
- Pandol SJ,Saluja AK,Imrie CW and Banks PA. Acute pancreatitis: bench to the bedside. Gastroenterology 132(3): 1127-1151,2007. PMID: 17383433.
- Perides G,Laukkarinen JM,Vassileva G and Steer ML. Biliary acute pancreatitis in mice is mediated by the G-protein-coupled cell surface bile acid receptor Gpbar1. Gastroenterology 138(2): 715-725,2010. PMID: 19900448.
- Pusl T,Vennegeerts T,Wimmer R,Denk GU,Beuers U and Rust C. Tauroursodeoxycholic acid reduces bile acid-induced apoptosis by modulation of AP-1. Biochem Biophys Res Commun 367(1): 208-212,2008. PMID: 18164257.
- Raraty M,Ward J,Erdemli G,Vaillant C,Neoptolemos JP,Sutton R, et al. Calcium-dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc Natl Acad Sci U S A 97(24): 13126-13131,2000. PMID: 11087863.
- Reber HA and Mosley JG. The effect of bile salts on the pancreatic duct mucosal barrier. Br J Surg 67(1): 59-62,1980. PMID: 7357247.
- Reber HA,Roberts C and Way LW. The pancreatic duct mucosal barrier. Am J Surg 137(1): 128-134,1979. PMID: 31807.
- Reber HA, Tweedie JH. Effects of a bile salt on the permeability of the pancreatic duct to macromolecules. Surg Forum 32: 219-221,1981.
- Renner IG and Wisner JR, Jr. Ceruletide-induced acute pancreatitis in the dog and its amelioration by exogenous secretin. Int J Pancreatol 1(1): 39-49,1986. PMID: 3693975.
- Renner IG,Wisner JR, Jr. and Rinderknecht H. Protective effects of exogenous secretin on ceruletide-induced acute pancreatitis in the rat. J Clin Invest 72(3): 1081-1092,1983. PMID: 6193140.
- Rodrigues CM,Fan G,Ma X,Kren BT and Steer CJ. A novel role for ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane perturbation. J Clin Invest 101(12): 2790-2799,1998. PMID: 9637713.
- Rodrigues CM,Fan G,Wong PY,Kren BT and Steer CJ. Ursodeoxycholic acid may inhibit deoxycholic acid-induced apoptosis by modulating mitochondrial transmembrane potential and reactive oxygen species production. Mol Med 4(3): 165-178,1998. PMID: 9562975.
- Saluja AK,Lerch MM,Phillips PA and Dudeja V. Why does pancreatic overstimulation cause pancreatitis? Annu Rev Physiol 69: 249-269,2007. PMID: 17059357.
- Senninger N. Bile-induced pancreatitis. Eur Surg Res 24 Suppl 1: 68-73,1992. PMID: 1601026.
- Swell L,Gustafsson J,Danielsson H,Schwartz CC,Halloran LG and Vlahcevic ZR. Bile acid synthesis in humans. Cancer Res 41(9 Pt 2): 3757-3758,1981. PMID: 7260942.
- Venglovecz V,Hegyi P,Rakonczay Z, Jr.,Tiszlavicz L,Nardi A,Grunnet M, et al. Pathophysiological relevance of apical large-conductance Ca(2)+-activated potassium channels in pancreatic duct epithelial cells. Gut 60(3): 361-369,2011. PMID: 20940280.
- Venglovecz V,Rakonczay Z, Jr.,Ozsvari B,Takacs T,Lonovics J,Varro A, et al. Effects of bile acids on pancreatic ductal bicarbonate secretion in guinea pig. Gut 57(8): 1102-1112,2008. PMID: 18303091.
- Voronina S,Longbottom R,Sutton R,Petersen OH and Tepikin A. Bile acids induce calcium signals in mouse pancreatic acinar cells: implications for bile-induced pancreatic pathology. J Physiol 540(Pt 1): 49-55,2002. PMID: 11927668.
- Voronina SG,Barrow SL,Gerasimenko OV,Petersen OH and Tepikin AV. Effects of secretagogues and bile acids on mitochondrial membrane potential of pancreatic acinar cells: comparison of different modes of evaluating DeltaPsim. J Biol Chem 279(26): 27327-27338,2004. PMID: 15084611.
- Voronina SG,Barrow SL,Simpson AW,Gerasimenko OV,da Silva Xavier G,Rutter GA, et al. Dynamic changes in cytosolic and mitochondrial ATP levels in pancreatic acinar cells. Gastroenterology 138(5): 1976-1987,2010. PMID: 20102715.
- Wen L,Voronina S,Javed MA,Awais M,Szatmary P,Latawiec D, et al. Inhibitors of ORAI1 Prevent Cytosolic Calcium-Associated Injury of Human Pancreatic Acinar Cells and Acute Pancreatitis in 3 Mouse Models. Gastroenterology 149(2): 481-492 e487,2015. PMID: 25917787.
- Zsembery A,Strazzabosco M and Graf J. Ca2+-activated Cl- channels can substitute for CFTR in stimulation of pancreatic duct bicarbonate secretion. FASEB J 14(14): 2345-2356,2000. PMID: 11053257.