
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2020.17
| Attachment | Size |
|---|---|
| 902.65 KB |
I. Introduction: Focus of the Chapter
The major cell types defining exocrine functions of pancreas are pancreatic acinar and pancreatic ductal cells. Acinar cells secrete digestive enzymes, precursors of digestive enzymes (zymogens) and NaCl-rich fluid, whilst ductal cells secrete large volumes of bicarbonate-containing fluid and form a conduit for exocrine pancreatic secretion (reviewed in (35, 76, 128)). Both exocytotic secretion of proteins and fluid secretion are energy consuming processes. Furthermore, pancreatic acinar cells have high levels of protein synthesis (165)) supporting substantial protein export by secretion. The bioenergetics of this organ are designed to sustain these energy demands and adjust ATP generation when cells undergo shifts between resting and stimulated conditions. Disruption/insufficiency of ATP generation is an important feature of pancreatic damage in pathological conditions. In this chapter we will consider both physiological adaptation of bioenergetics in exocrine pancreas and its disruption in acute pancreatitis.
Pancreatic acinar cells constitute more than 80% of the volume of the exocrine pancreas (24) and are, thus, likely to make major contributions to bioenergetics parameters measured in experiments utilizing undissociated pancreas. Signaling mechanisms and bioenergetics of this cell type have been extensively investigated and a substantial part of this chapter therefore will be focused on the bioenergetics of pancreatic acinar cells. There has also been recently significant progress in characterizing bioenergetics changes in ductal cells, particularly in conjunction with the pathophysiology of acute pancreatitis; we will discuss this progress in section ‘Alteration of Bioenergetics in Pancreatic Pathophysiology’.
II. Mechanisms of ATP Generation
Information on the relative importance of different substrates (carbohydrates, amino acids and fatty acids) for the bioenergetics of exocrine pancreas is somewhat limited. Pancreatic acinar cells express a range of amino acid transporters (144), allowing accumulation of amino acids against a significant concentration gradient; this is beneficial for prominent protein synthesis in this cell type and could also support utilisation of amino acids as substrates for ATP production. Notably, in in vivo and in vitro experimental models of acute pancreatitis expression of several amino acid transporters was significantly downregulated (144). A study by S. Araya and colleagues highlighted a high rate of glutamine transport in pancreatic acinar cells and compared CO2 generation in response to addition of amino acids (glutamine and leucine), glucose and palmitoyl (3). The study concluded that glutamine is the preferred energy source for pancreatic acinar cells and that these cells operate an efficient conversion of glutamine to glutamate followed by glutamate utilisation (via α-ketoglutarate) in the Krebs cycle (3). Information on the contribution of other amino acids and fatty acids, however, is sparse. Glucose can support the bioenergetics of acinar cells and glucose-containing extracellular solution has been frequently utilized in studies of glycolysis and oxidative phosphorylation in this cell type. The relative contribution of oxidative metabolism and glycolysis to the net ATP generation in exocrine pancreas and pancreatic acinar cells is still a subject for debate. Mitochondria occupy a significant proportion of cell volume in pancreatic acinar cells (approximately 8 % (24)). An early study by Bauduin and colleagues concluded that mitochondria are by far the major source of ATP production in acinar cells whilst glycolysis makes only minor contribution (14). This paper, however, reported that high concentration of Oligomycin (inhibitor of the mitochondrial F1F0-ATP synthase: abbreviated to ATP synthase) reduced nucleoside triphosphate (presumably mainly ATP) content to approximately 41% of the control value, suggesting a significant contribution of glycolysis to ATP generation. However, the same authors reported that antimycin (inhibitor of the mitochondrial electron transport chain) reduced nucleoside triphosphate content to 16% and anaerobiosis to 8% (14). H. Kosowski and colleagues from W. Halangk laboratory reported a decrease of ATP content to approximately 21% of the control value following 30 minuses of anoxia (98). Experiments with anoxia and antimycin could be interpreted as evidence for a rather modest contribution of glycolysis to ATP generation in the acinar cells. However, these treatments are expected to trigger the reverse mode of ATP synthase, which can convert this enzyme into a powerful ATP consumer (i.e. in this mode the enzyme functions as an ATPase consuming ATP and transporting protons into the matrix; reviewed in (32, 69, 101, 167)). The discrepancy between the ATP levels retained in the presence of oligomycin and after the inhibition of mitochondrial electron transport chain by anoxia or antimycin could be explained by a reverse mode of ATP synthase. ATP consumption by this reverse mode could be potentially prevented by the F1F0-ATP synthase inhibitory factor 1 (IF1) (32). However, it should be noted that normal pancreas contains relatively low levels of IF1 (e.g. in comparison with heart or pancreatic cancer tissue (158)) and therefore the effect of the reverse mode could be significant. Consequently, experiments with oligomycin or combination of oligomycin with an inhibitor of the electron transport chain should provide a better estimation of contribution made by glycolysis to ATP generation of the acinar cells. The early estimation of 41% (14) is consistent with more recent findings indicating a significant (at least 30-40%) contribution of glycolysis to ATP generation in acinar cells (158, 171). Furthermore, the balance of glycolytic and oxidative ATP production depends on external factors. In particular, secretagogues that produce moderate Ca2+ signals upregulate the rate of Krebs cycle and mitochondrial ATP generation ((39, 170, 171) and the next section), whilst application of insulin strongly potentiates glycolytic ATP production, which can sustain bioenergetics of acinar cells with damaged mitochondria (111, 145).
III. Stimulus-Metabolism Coupling
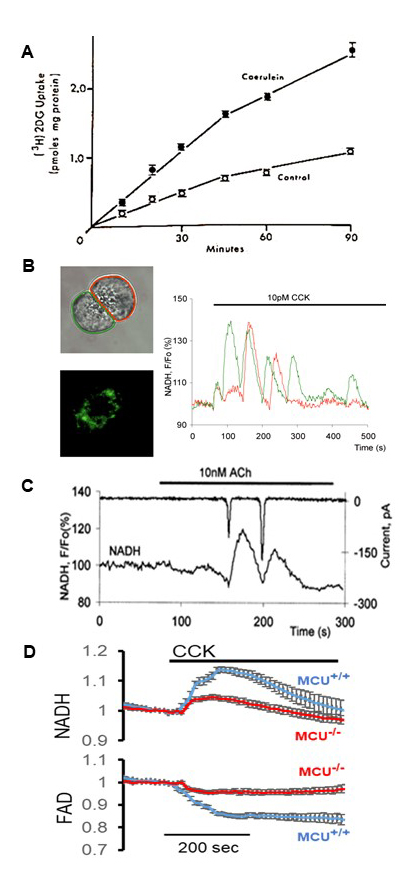
In the late 1970s,an important and surprising observation was made in experiments on isolated pancreatic acini (97, 177); upon stimulation with secretagogues (caerulein, cholecystokinin or carbachol) cellular ATP levels were stable or even slightly increased (97, 177). It would be expected that the increased energy demands imposed by the stimulation of secretion should reduce ATP content. The findings were therefore paradoxical and suggested existence of an effective mechanism(s), capable of compensating and even overcompensating for the increased ATP usage in the stimulated cells i.e. indicative of an efficient stimulus-metabolism coupling in this cell type. Early evidence for such stimulus-dependent upregulation of bioenergetics came from the study by M. Korc and colleagues demonstrating strong and rapid upregulation of glucose uptake in isolated pancreatic mouse acini stimulated with secretagogues ((97); see Figure 1A). Caerulein, CCK and Carbachol were tested and all produced clear and substantial increases in glucose uptake (2.5-3 times in comparison with control) (97). Crucially, the increase in the glucose uptake could be mimicked by the Ca2+ ionophore A23187. Furthermore, incubation of cells in the solution containing the Ca2+ chelator EGTA suppressed the agonist-induced glucose uptake (97). The authors therefore concluded that intracellular Ca2+ may “…mediate hormone-stimulated glucose transport…” (97). The authors’ suggested role of cellular Ca2+ in the regulation of bioenergetics of the acinar cells was confirmed by the following 40 years of research. Important findings that characterized the role played by Ca2+ signaling in regulating bioenergetics were made at the end of the previous century in Denton’s laboratory. These experiments revealed the ability of Ca2+ to upregulate the rate of Krebs cycle; the effect of Ca2+ was mediated by rate-limiting dehydrogenases of the Krebs cycle (reviewed in (53, 119)). At the single cell level, the relationships between cytosolic Ca2+, mitochondrial Ca2+ and NAD(P)H (an important reducing equivalent produced in Krebs cycle that supplies electrons for the respiratory chain) were first demonstrated in hepatocytes (69). This study utilized cellular autofluorescence to monitor the NAD(P)H responses and revealed fast upregulation of the NAD(P)H production by Ca2+ transients (69). In isolated pancreatic acinar cells relationships between Ca2+ signaling (in cytosol and mitochondria) and NAD(P)H was first characterized by Voronina and colleagues (170). This study revealed fast and robust NAD(P)H transients associated with Ca2+ responses (Figure 1B and C), consistent with NAD(P)H responses recorded from perfused pancreas (87).
Subsequently, rises of NAD(P)H in response to CCK-induced Ca2+ transients were shown to occur in isolated human pancreatic acinar cells, confirming a similar stimulus-metabolism coupling to that previously elucidated in murine cells (125). Physiological concentrations of important secretagogues (e.g. CCK and ACh) induce oscillatory Ca2+ responses in acinar cells (reviewed in (137, 176, 179)). NAD(P)H responses were sufficiently fast to forms oscillations (reflecting oscillations in the rate of the Krebs cycle) closely associated with the oscillations of cytosolic Ca2+ (170)). Upon stimulation with Ca2+ releasing secretagogues pancreatic acinar cells generate local and global Ca2+ transients (e.g. (90, 160) reviewed in (137, 179)). All global Ca2+ transients were able to trigger NAD(P)H transients (i.e. increased rate of the Krebs cycle). The majority of local Ca2+ responses were also able to trigger resolvable NAD(P)H responses except for the very short local Ca2+ transients (less than 2.9 seconds) which are probably quenched by cytosolic calcium buffers before reaching the mitochondria (170). To upregulate activity of dehydrogenases of the Krebs cycle and accelerate its rate, Ca2+ must enter mitochondrial matrix. The phenomenon and properties of mitochondrial Ca2+ entry were extensively characterized in the second part of the previous century; however, the molecular mechanism was only discovered in 2011.
Figure 1. Stimulus-metabolism coupling in pancreatic acinar cells. (A) Changes in glucose uptake by pancreatic acini upon application of caerulein. (Adapted from Korc et al (97)). (B) NADH oscillations in pancreatic acinar cells. Left panels show transmitted image of two pancreatic acinar cells and regions of interest for recording of fluorescence (corresponding to traces on the right panel). Lower left panel shows the distribution of NADH fluorescence. (Adapted from Voronina et al (170)). (C) Correlation between cytosolic Ca2+ responses (revealed by negative deflections on the trace of Ca2+-dependent Cl- current) and changes of NADH fluorescence. (Adapted from Voronina et al (170)). (D) Effect of mitochondrial Ca2+ uniporter (MCU) knockout on the NADH and FAD responses to CCK stimulation in pancreatic acinar cells. (Adapted from Chvanov et al (39)).
Mitochondrial Ca2+ entry is primarily mediated by the mitochondrial Ca2+ uniporter complex ((15, 52) reviewed in (110)). In mammals this complex is composed of the mitochondrial Ca2+ uniporter (MCU, pore forming protein localized in the inner mitochondrial membrane), its Ca2+ dependent regulators MICU1/MICU2 (47, 86, 132, 135), and other associated and regulatory proteins (reviewed in (110)). An important function of MICU1/MICU2 is to provide a threshold Ca2+ concentration that limits Ca2+ entry into mitochondria at low cytosolic Ca2+ concentration. The voltage difference between mitochondria and cytosol is substantial (approximately 140 mV); it is sufficient to drive Ca2+ entry into mitochondria even at low resting Ca2+ levels. This would form a futile Ca2+ cycle necessitating continuous energy dependent Ca2+ extrusion from mitochondria (20, 21, 47, 86, 109, 132). At higher Ca2+ concentrations MICU1 and MICU2 bind Ca2+ and undergo conformational change allowing Ca2+ entry through the MCU channel (47, 86, 109, 132). Threshold Ca2+ concentrations necessary for such ‘opening’ of the MCU complex are usually significantly higher than the resting cytosolic Ca2+ and usually also higher than Ca2+ concentrations in the areas distant from Ca2+ releasing channels in the cellular organelles or Ca2+ influx channels in the plasma membrane. Therefore Ca2+ entry into mitochondrial matrix in many systems depends on the mitochondrial localisation and specifically on the proximity of the organelles and their MCU complex to the Ca2+ releasing channels (reviewed in (159)). In pancreatic acinar cells mitochondria are found in three major groups - subplasmalemmal, perigranular and perinuclear (54, 83, 130, 161). These mitochondria are conveniently localized to respond to Ca2+ release from IP3 receptors (IP3Rs) (161), Ryanodine Receptors (RyR) (156) and store operated Ca2+ (SOC) influx channels (103, 130). The mitochondrial Ca2+ uniporter is responsible for the bulk of Ca2+ entry into the mitochondrial matrix, however, other mitochondrial Ca2+ influx mechanisms have been reported (e.g. (25, 72, 147)). A recent study, utilizing MCU knockout (MCU KO) mice, demonstrated that the MCU complex is responsible for the major component of the mitochondrial Ca2+ entry in CCK-stimulated pancreatic acinar cells, however, very small mitochondrial Ca2+ signals were observed in CCK-stimulated acinar cells from MCU KO mice suggesting an alternative (and currently unknown) Ca2+ entry mechanism in pancreatic mitochondria (39). These observations were consistent with the results of measurements of mitochondrial NAD(P)H and FAD responses. During the Krebs cycle FAD is converted to FADH2; FAD is fluorescent and the decrease of its content/fluorescence in mitochondria suggests its reduction and could be interpreted (with caution) as an acceleration of Krebs cycle (57). NAD(P)H and FAD responses associated with Ca2+ signals have been measured in a number of cell types (e.g. (56, 69)); in these studies, transient increases in NAD(P)H fluorescence were usually accompanied by decreases in FAD fluorescence. The amplitudes of both NAD(P)H and FAD responses to CCK stimulation drastically decreased in the acinar cells from MCU KO animals in comparison with wild type (WT) mice ((39) and Figure 1D). This confirms the major role of Ca2+ signals and MCU-mediated mitochondrial Ca2+ responses in stimulus-metabolism coupling in pancreatic acinar cells. Notably small NAD(P)H and FAD responses were retained in the cells from MCU KO mice; this is consistent with the retention of small mitochondrial Ca2+ signals in cells of these animals (39). Another important parameter reflecting cellular bioenergetics is oxygen consumption. In line with the observed upregulation of glucose uptake (98) and NAD(P)H increase (39, 170), oxygen consumption significantly increases in perfused pancreas (87) and isolated acinar cells stimulated with Ca2+ releasing secretagogues (39, 87, 112, 121). Importantly, this effect was diminished in acinar cells from MCU KO mice (39), again highlighting the role of mitochondrial Ca2+ in the stimulus-metabolism coupling of this cell type. The notion of the efficient stimulus-metabolism coupling in pancreatic acinar cells is consistent with the outcome of experiments involving ATP and creatine phosphate measurements in perfused rat pancreas, which indicated that “ATP and PCr remained largely unchanged” in spite of maximal stimulation of the organ with 100 pM CCK producing robust protein and fluid secretion (117). In this study a higher concentration of CCK (1nM) produced a small decrease in ATP levels, which may be associated with tissue-level effects of supramaximal doses of this agonist and will be discussed in the role of bioenergetics in pancreatitis (see below). Whilst studies of the relationships between Ca2+-releasing secretagogues and bioenergetics, conducted on isolated acinar cells and perfused pancreata, reported stable or minimally changed ATP levels, despite upregulation of energy consuming processes (97, 171, 177); results from experimental animal models, involving low “physiological” as well as supramaximal doses of secretagogues, are somewhat contradictory. In the study by R. Luthen and colleagues, all tested concentrations of caerulein (7 hourly intraperitoneal injections of caerulein each corresponding to 0.1, 1, 5 and 50 µg/kg body weight) produced significant and similar decreases in ATP concentration in mouse pancreata (by approximately 33% one hour after the first injection and 45% one hour after the last injection)(104). It is conceivable that in intact tissue the rate of ATP production is limited by tissue oxygen levels and nutrient supply (e.g. due to changes of pancreatic microcirculation, reviewed in (48)), which are usually not restricted in single cell preparations. It should be also noted that other studies involving animal models and stimulation with secretagogues reported stable pancreatic ATP levels (117, 127). This apparent contradiction emphasizes the importance of continuing investigation of stimulus-metabolism coupling at cellular and tissue levels as well as in animal models. The pathophysiology of acute pancreatitis is frequently associated with disruption of bioenergetics and loss of ATP in pancreatic tissue. In experimental models of acute pancreatitis this is particularly clear for bile-induced and fatty acid-induced acute pancreatitis; the relevant studies will be discussed below.
IV. Metabolism - Signaling Feedback
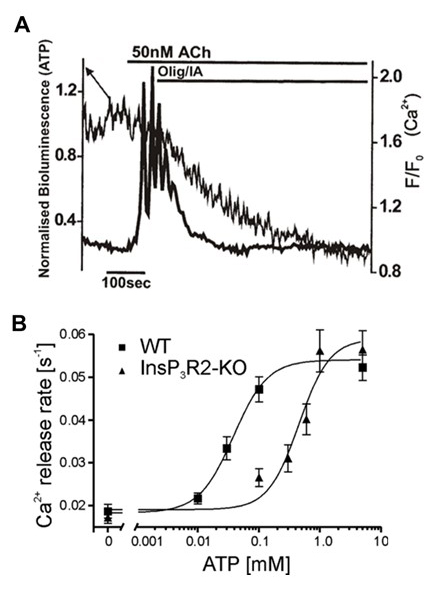
Several pathophysiologically-relevant substances (including fatty acids, reactive oxygen species, asparaginase, bile acids and non-oxidative alcohol metabolites) can deplete cellular ATP ((5, 26, 44, 111, 133, 134, 145, 171); and the next section). While Ca2+ signaling can influence metabolism and mediate efficient stimulus-metabolism coupling in pancreatic acinar cells, there is also a powerful effect of metabolism on signaling. Interesting early manifestation of such feedback mechanism came from study by A. Hofer and colleagues which demonstrated inhibition of Ca2+ leak from the ER lumen by ATP depletion (78). Further studies identified the prominent influence of ATP depletion on ACh-induced Ca2+ oscillations in pancreatic acinar cells (12); a modest decrease in cytosolic ATP concentration (revealed by measuring luciferase bioluminescence in live cells) resulted in abrupt termination of Ca2+ oscillations ((12) and Figure 2A). The molecular phenomenon underlying this cellular response was identified in studies from the Yule laboratory that characterized ATP sensitivity of IP3 receptors responsible for the Ca2+ oscillations in the acinar cells. All three types of IP3Rs are expressed in pancreatic acinar cells (62), however, the expression of InsP3R1 is relatively low (63) and it is unlikely to play significant role in physiologically-relevant Ca2+ responses. InsP3R2 and InsP3R3 are the major receptor subtypes in acinar cells (62). These are ATP sensitive and Ca2+ fluxes via these channels are inhibited by ATP reduction in mM or sub-mM range (18, 129). Further study utilizing acinar cells from InsP3R2 knock-out mice revealed particular importance of this Ca2+ releasing channel in determining ATP sensitivity of Ca2+ responses ((129) and Figure 2B). Another vital component of calcium signaling, Ca2+ influx via the plasma membrane, is also ATP-sensitive. In pancreatic acinar cells, Ca2+ influx is primarily mediated by the store operated Ca2+ entry (SOCE) based on the interaction of the ER Ca2+ sensor STIM1 and the plasma membrane channel-forming protein Orai (1, 64, 103, 168, 172). A role for TRPC channels in SOCE has been also reported (92).
Figure 2. Metabolism-signaling feedback. (A) Inhibition of acetylcholine induced cytosolic Ca2+ oscillations in a pancreatic acinar cell by ATP depletion (induced by application of oligomycin and iodoacetate). (Adapted from Barrow et al (12). (B) ATP sensitivity of Ca2+ release from internal stores of InsP3 stimulated permeabilized pancreatic acinar cells. The figure compares ATP sensitivity of cells isolated from wild type mice (WT) and from mice with knocked out InsP3R2. The figure was adapted with modifications from Park HS et al. The type 2 inositol (1,4,5)-trisphosphate (InsP3) receptor determines the sensitivity of InsP3-induced Ca2+ release to ATP in pancreatic acinar cells (129).
ATPdepletion resulted in the efficient inhibition of the store operated Ca2+ entry (SOCE) in pancreatic acinar cells (12). Inhibition of mitochondria and/or ATP depletion have been shown to suppress SOCE entry in a number of other cell types (63, 66, 79, 114, 146) and could be therefore considered as a general phenomenon not limited to the pancreatic acinar cell. The specific molecular mechanism responsible for such inhibition is still debated. Stimulus-metabolism coupling and metabolism-signaling feedback are schematically illustrated in Figure 3. As expected, ATP depletion also suppressed Ca2+ uptake into the ER lumen via sarcoplasmic / endoplasmic reticulum Ca2+ ATPase (SERCA) and Ca2+ extrusion via plasma membrane Ca2+ ATPase (PMCA) (12, 111, 145). In pancreatic acinar cells inhibition of SOCE by ATP depletion occurred in parallel with the inhibition of Ca2+ extrusion (12) suggesting an important and hitherto unidentified link between the two processes. The inhibition of Ca2+ influx by ATP depletion is, however, incomplete and prolonged depletion leads to Ca2+ overload and damage/death of pancreatic acinar cells (111, 145). Furthermore, some inducers of acute pancreatitis (e.g. bile acids and fatty acids) seem to be capable of generating an alternative Ca2+ influx pathway which is not inhibited by ATP depletion (12, 111, 145). A number of novel Ca2+ influx channels have been recently identified in pancreatic acinar cells (60, 143, 157) and the effects of mitochondrial inhibition and ATP depletion on these channels is currently unknown. Identifying the relationships between the recently identified Ca2+ entry pathways and cellular bioenergetics could be the subject of productive future investigation.
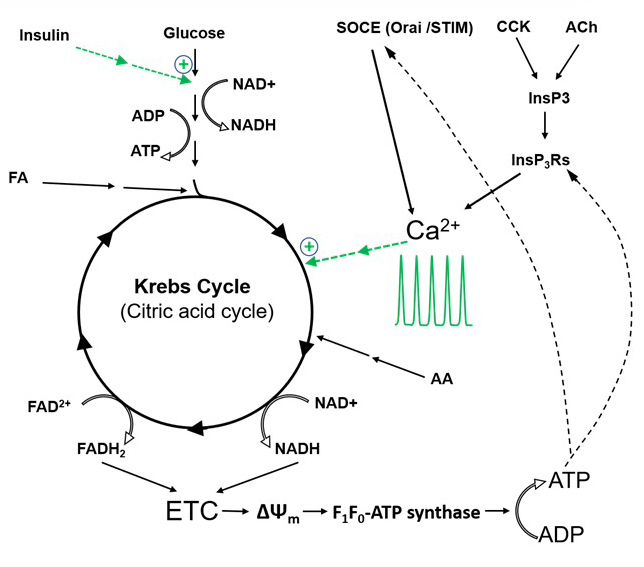
Figure 3. Simplified diagram of stimulus-metabolism coupling and metabolism-signaling feedback in pancreatic acinar cells. The schematic illustrates the mechanisms of physiological adaptation of bioenergetics to modulate stimulation by secretagogues. In the diagram ΔΨm indicates mitochondrial membrane potential (major component of the proton-motive force), ETC – electron transport chain, AA - amino acids, FA - fatty acids. Dashed green lines illustrate the stimulating effect of physiological Ca2+ signals (Ca2+ oscillations induced by ACh or CCK) on the Krebs cycle and of insulin on glycolysis. Dashed black lines highlight the role of ATP in the regulation of InsP3 receptors (InsP3Rs) and store operated Ca2+ entry (SOCE). Both IP3Rs and the SOCE pathway are ATP-sensitive mechanisms. In resting unstimulated cells and those stimulated by physiological concentrations of secretagogues, ATP levels are sufficient to sustain both InsP3R -mediated Ca2+ release from the endoplasmic reticulum and SOCE via the STIM/Orai mechanism. However, ATP depletion (e.g. triggered by the inducers of acute pancreatitis) can suppress the InsP3R -mediated Ca2+ release and SOCE. This is also accompanied by the inhibition of Ca2+ leak from the ER, Ca2+ uptake into the ER by sarcoplasmic/endoplasmic reticulum Ca2+ pumps and Ca2+ extrusion by Ca2+ pumps of the plasma membrane (not shown). - For the sake of clarity/simplicity we have omitted some Ca2+-mobilizing second messengers and corresponding intracellular Ca2+-releasing channels. (For detailed reviews of Ca2+ signaling in this cell type, see Refs. (179) and (137)). We have also simplified the entry mechanisms of amino acid (AA) and fatty acid (FA) derivatives into the Krebs cycle. Feedback regulation of the rate of the Krebs cycle by ATP, ADP and NAD(P)H was also omitted on this diagram -.
The pronounced effect of cellular metabolism on Ca2+ signaling limits energy expenditure associated with actual signaling responses and downstream reactions of this critical for the acinar cells signaling cascade. It also offers some protection against Ca2+ overload and associated Ca2+ toxicity. This protection is however incomplete and can be overwhelmed by the inducers of acute pancreatitis.
V. Alteration of Bioenergetics in Pancreatic Pathophysiology
Ca2+-dependent mitochondrial dysfunction is a core feature of acute pancreatitis. In contrast to the spatially restricted, oscillatory Ca2+ signaling events that occur in response to physiological levels of secretagogues in isolated pancreatic acinar cells, supramaximal stimulation causes damaging global, sustained calcium elevations (136, 141, 173). The ability to cause overload of cytosolic Ca2+ and thereby disrupt Ca2+ homeostasis in acinar cells is a principal pathological feature common to known precipitants of acute pancreatitis, including bile acids, non-oxidative ethanol metabolites and fatty acids (46, 92) (see Figure 4A), and is an important trigger for mitochondrial damage that compromises cellular ATP production (45). The importance of altered energy levels within the pancreas relevant to its pathology has been recognized for many years, although in vivo studies have produced somewhat varied results. In the early 1990s, substantial falls of cellular ATP in the pancreas were detected in experimental AP models (using non-invasive NMR spectrometry of 31P) (127). Interestingly, there was heterogeneity in responses dependent on the model employed. Thus, whereas significant falls of ATP were detected in AP induced by fatty acid (oleic acid) infusion or regional ischemia, levels were unchanged in caerulein hyperstimulation and partial ductal obstruction models. In contrast, a contemporaneous study in rat pancreas showed that ATP levels were more than halved from controls in the caerulein AP model (77). A subsequent investigation by W. Halangk and colleagues supported these findings, demonstrating a reduction of phosphorylating respiration in acinar cells and fall of ATP to 57% in caerulein AP after 24 hours (using a Clark-type electrode) (70). The authors concluded that caerulein-induced acute pancreatitis is accompanied by “…a drastic and long-lasting reduction of the capacity for mitochondrial ATP production…”. It should be noted that this phenomenon developed relatively slowly i.e. at early time points (up to 5 hours) the cellular ATP content reported was not significantly diminished from controls and therefore slower than other manifestations of experimental acute pancreatitis like vacuolisation (37) and intracellular trypsinogen activation (108). In contrast, a recent study found that high levels of L-lysine caused mitochondrial damage which occurred before activation of trypsinogen and NFkB, potentially via impairment of electron transport chain coupling or a disbalance between hydrolysis and synthesis of ATP (22). To-date, the balance of evidence from in vivo models has demonstrated loss of pancreatic ATP as acute pancreatitis develops, indicating that early mitochondrial damage is a critically important factor in disease progression; variability between studies may reflect methodological differences, etiology and/or severity of the experimental model and further comparative studies are warranted. The in vivo findings are, therefore, consistent with ATP measurements on isolated acinar cells showing rapid declines of ATP concentration in cells treated with fatty acid (POA) or bile acid (TLC-S) (45, 171). In addition, the bile acid chenodeoxycholate has been shown to decrease intracellular ATP levels in pancreatic ductal cells, with inhibition of both oxidative and glycolytic metabolism linked to impaired bicarbonate secretion (91, 107). With respect to in vivo models, the bioenergetics of acinar and ductal cells in pancreatic tissue will be influenced by changes in local blood flow. This is particularly relevant to the pathophysiology of acute pancreatitis, which is associated with impairment of microvascular perfusion (e.g. (120, 139, 148, 174) reviewed in (48)). Inducers of acute pancreatitis have been shown to increase the permeability of pancreatic capillaries (e.g. (120, 139, 148), reduce microvascular blood flow (162) and pancreatic oxygen tension (93, 162)). Notably, different models of experimental acute pancreatitis are associated with distinct changes in the microcirculation, ranging from minimal responses and even modest increases of capillary perfusion in mild acute pancreatitis (95) to drastic decreases of capillary perfusion associated with more severe models (95, 162). The intrinsic abilities of the exocrine cells to adjust metabolism and signaling in response to secretagogues and the inducers of acute pancreatitis (discussed in the previous section) will be therefore “superimposed” on changes in oxygen supply and nutrient availability determined by the microcirculation.
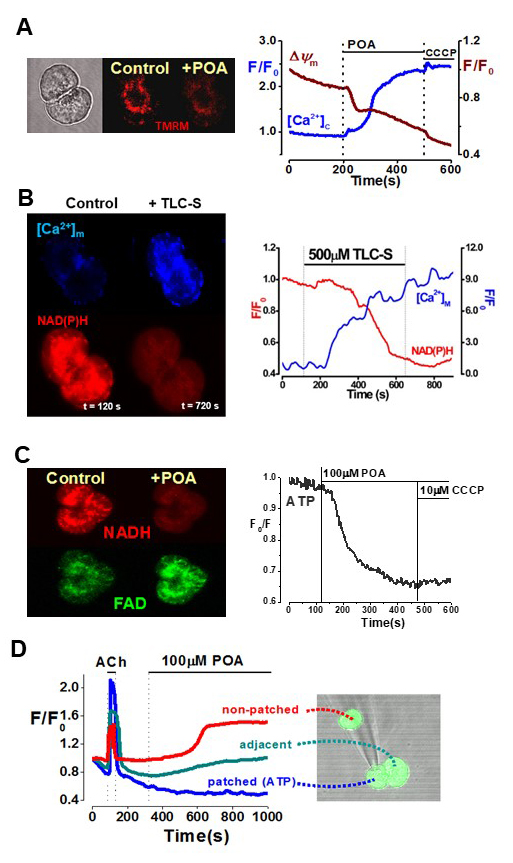
The mechanisms underlying ATP depletion induced by acute pancreatitis precipitants have been addressed in detail using isolated pancreatic acinar cells. Confocal microscopy investigations demonstrated that Ca2+ overload of acinar cells caused rapid depolarization of the mitochondrial membrane potential, accompanied by decreased NAD(P)H and increased FAD+ levels, with a resultant cellular ATP fall leading to necrosis. When applied at lower concentration (200 µM) the bile acid TLCS caused predominantly oscillatory calcium rises, accompanied by NADH elevations, in accord with stimulus-metabolism coupling (as described above). However, sustained Ca2+ elevations at higher concentration (500 µM) led to rises of mitochondrial Ca2+ with dramatic decrease of NAD(P)H (26, 169) (see Figure 4B). Similarly, the non-oxidative ethanol metabolite palmitoleic acid ethyl ester (POAEE) and POA, produced by hydrolysis of the fatty acid ethyl ester (53, 80), induced sustained elevations of cytosolic Ca2+ that resulted in mitochondrial dysfunction (45). Simultaneous measurements of cytosolic Ca2+ and Δψm revealed the temporal relationship of these events; as cytosolic Ca2+ progressively rose there was a mirrored mitochondrial depolarization and rapid decrease of NAD(P)H (42) (see Figure 4A). The consequence of mitochondrial impairment was a profound depletion of intracellular ATP (measured using with Magnesium Green); this effect was maximal and not further increased by the protonophore CCCP (45) (see Figure 4C). Interestingly, a Ca2+-independent action of POA was also detected, revealed by pre-treatment with the Ca2+ chelator BAPTA which prevented the loss of Δψm although NAD(P)H decrease still persisted. The rundown of mitochondrial ATP production has dramatic effects on Ca2+ homeostasis in the pancreatic acinar cell, which appears particularly vulnerable to sustained cytosolic Ca2+ increases; unlike excitable cells such as cardiomyocytes, the contribution of the Na+/Ca2+ exchanger to Ca2+ homeostasis in the pancreatic acinar cell appears weak or absent (123). The major routes for Ca2+ clearance, the SERCA and PMCA pumps, are fuelled by cellular ATP and would be progressively impaired as energy levels fall during acute pancreatitis development (11, 29). This is likely to result in a vicious cycle in which Ca2+ dependent mitochondrial dysfunction perpetuates sustained cytosolic Ca2+ elevation, resulting in augmented necrotic cell death (43). The importance of maintaining ATP levels for acinar cell function was demonstrated by the provision of intracellular ATP via a patch pipette (45); this prevented the damaging Ca2+ elevations induced by POA, confirming the crucial roles of the SERCA/PMCA for Ca2+ clearance in pancreatic acinar cells (45) (see Figure 4D). In accord, ATP provision prevented the sustained Ca2+ rises and consequent acinar cell necrosis induced by bile acid (26), while supplemental ATP was able to mitigate the detrimental effects of ethanol and its non-oxidative metabolites on CFTR in ductal cells that promote acute pancreatitis (84, 105).
Figure 4. Pathological Ca2+-dependent mitochondrial bioenergetic dysfunction in pancreatic acinar cells. (A) Correlation between palmitoleic acid (POA)-induced sustained cytosolic Ca2+ response (Fluo4) and mitochondrial depolarization (TMRM). (Adapted from Criddle et al. (45)). (B) Correlation between bile acid (TLCS: taurolithocholic acid sulphate)-induced sustained mitochondrial Ca2+ rises (Rhod2) and decrease of NAD(P)H (autofluorescence) in a doublet of pancreatic acinar cells (left panel) and corresponding traces (right panel). (Adapted from Booth et al. (26)). (C) Bioenergetics changes in response to POA in pancreatic acinar cells, shown as concurrent decrease of mitochondrial NADH and increase of FAD (autofluorescence: left panel) and depletion of ATP (Magnesium: Green). (D) Provision of ATP via a patch-pipette prevents toxic POA-induced sustained Ca2+ elevation in pancreatic acinar cells, highlighting the vital importance of ATP-dependent Ca2+ pumps to Ca2+ clearance and prevention of cell damage (C, D). (Adapted from Criddle et al. (45)).
A. How does ATP depletion occur?
The precise mechanisms of ATP depletion are still unclear, although a crucial role of MPTP opening in mediating mitochondrial dysfunction has been identified in the pancreas (23, 124, 152), in common with other tissues such as heart, liver and brain (30, 89, 153). The formation of this mega-pore causes a profound permeabilization of the inner mitochondrial membrane that allows movement of solutes <1.5kD in and out of the organelle. Although the exact composition of the pore is contentious and currently unresolved (17, 31, 73, 74, 88), prevention of MPTP opening is achieved via inhibition of the modulatory protein cyclophilin D (CypD); this protein is therefore considered an important target for acute pancreatitis therapy (40, 82, 124, 150, 163). Our group has shown that knockout and pharmacological inhibition of CypD ameliorated damage in isolated murine and human acinar cells induced by acute pancreatitis precipitants and was protective in multiple in vivo experimental models reflecting the principal etiologies (including biliary: TLCS-AP (100) and alcoholic: FAEE-AP (80)) (124). Furthermore, CypD-sensitive MPTP formation mediated mitochondrial dysfunction in pancreatic ductal cells, indicating a common mechanism operating in multiple cell types implicated in the pathogenesis of acute pancreatitis (76, 163). The principal trigger for MPTP opening is the sustained elevation of mitochondrial Ca2+ (65), however, reactive oxygen species (ROS) have also been implicated in triggering MPTP channel formation (16, 59). In human and murine pancreatic acinar cells bile acid-induced effects are complex since sustained elevations of cytosolic and mitochondrial Ca2+ lead to sustained ROS increases; both Ca2+ and ROS formation are therefore likely to be important to the outcome of bioenergetics changes that underlie mitochondrial dysfunction in acute pancreatitis (26). Physiological and pathophysiological changes of acinar cell bioenergetics are schematically illustrated in Figure 5.
Figure 5. Diagram illustrating the effects of physiological and pathophysiological Ca2+ signals on mitochondrial bioenergetics that determine pancreatic acinar cell responses and fate. Physiological stimulation of pancreatic acinar cells with secretagogues cholecystokinin (CCK) and acetylcholine (ACh) elicits oscillatory Ca2+ signals that stimulate mitochondrial ATP production (stimulus-metabolism coupling) that fuels secretion. However, when subjected to stress, cellular bioenergetics are perturbed leading to instigation of cell death patterns. Precipitants of acute pancreatitis (AP), including bile acids, non-oxidative ethanol metabolites (fatty acid ethyl esters: FAEEs), fatty acids (FAs) and secretagogue hyperstimulation, cause sustained cytosolic Ca2+ elevations raise mitochondrial matrix Ca2+. Ca2+-dependent ROS generation in the mitochondria preferentially promotes apoptotic cell death, which may constitute a local protective mechanism whereby necrosis is avoided. However, as the level and/or duration of stress increases, a shift from apoptotic to necrotic cell death ensues. Ca2+-dependent formation of the mitochondrial permeability transition pore (MPTP) is a primary event that results in mitochondrial depolarization, altered NADH/FAD levels, ATP fall and necrotic cell death development. Elevated ROS levels also drive necrosis in a manner that is independent of the MPTP. Since clearance of cytosolic Ca2+ from the acinar cell is acutely dependent on ATP-requiring pumps (plasmalemmal Ca2+-ATPase (PMCA) and sarco-endoplasmic reticulum Ca2+-ATPase (SERCA)), the depletion of ATP evokes a vicious cycle which perpetuates Ca2+ overload and mitochondrial dysfunction. (Adapted from Criddle, DN 41)).
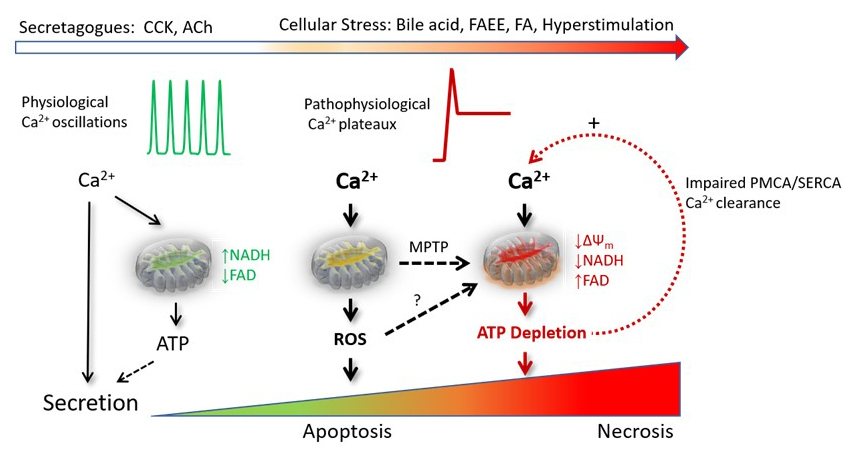
B. Role of ROS in bioenergetic alterations
Secretagogues and bile acids increase ROS generation in pancreatic acinar cells (26, 38); reviewed in (41)). In pancreatic acinar cells augmented ROS production promotes apoptosis (44), which may be an endogenous protective mechanism that enables the cell to deal with cellular stress by avoiding necrosis (19, 26, 44, 85). Necrosis is considered a largely uncontrolled form of cell death and in acute pancreatitis is associated with development of systemic inflammation (94); accordingly, inhibition of caspases caused a more severe necrotizing pancreatitis (19, 113). Our recent work has demonstrated that oxidative stress induced by exogenous H2O2 or menadione, which generates intracellular ROS via redox cycling (44), caused bioenergetics changes that were independent of CypD-sensitive MPTP opening (6). Increasing levels of oxidants elicited an apoptosis to necrosis shift that was unaffected by knockout of CypD or pharmacological inhibition with cyclosporin A. This lack of sensitivity to CypD is consistent with evidence in primary hepatocytes showing that CypD deletion affected MPTP formation induced by Ca2+ but not by thiol oxidants (13) and a model in which there is a regulated (Ca2+ activated and cyclosporin-sensitive) and unregulated (cyclosporin-insensitive) MPTP (8, 75). Therefore, counteracting excessive ROS in the pancreas with antioxidants might be desirable. However, despite many clinical trials for acute pancreatitis, results have generally been disappointing or inconclusive (4). A more recent approach to combat oxidative stress is the targeting of antioxidants to mitochondria in order to produce more localized, specific actions (126). For example, MitoQ, which accumulates in mitochondria due to its positively charged triphosphonium ion (TPP+) (10), prevented ROS-mediated damage in diverse cell types (33, 36, 49, 50, 102), although efficacy in clinical investigations so far has been disappointing (140, 156). In isolated acinar cells MitoQ reduced H2O2-mediated ROS elevations yet afforded no protection against loss of mitochondrial membrane potential induced by CCK or bile acid (81). Furthermore, MitoQ treatment produced mixed effects in in vivo acute pancreatitis models. When applied as a treatment after disease induction, it was unprotective in TLCS-AP but caused partial amelioration of histopathology scores in CER-AP, albeit without concomitant reduction of biochemical parameters (pancreatic trypsin and serum amylase). Interestingly, lung myeloperoxidase and interleukin-6 were concurrently increased by MitoQ in CER-AP, which may involve biphasic effects on ROS in isolated polymorphonuclear leukocytes, inhibiting acute increases but causing elevation at later time points. In pancreatic acinar cells, MitoQ exerted complex actions on mitochondrial bioenergetics that may in part mediated by the targeting TPP+ moiety (5). Thus, MitoQ and the general antioxidant n-acetyl cysteine caused sustained elevations of basal respiration and inhibition of spare respiratory capacity, an important indicator of the cell’s ability to respond to stress (149, 178), actions attributable to an antioxidant action. However, mitochondrial ATP turnover capacity and cellular ATP concentrations were also markedly reduced by both MitoQ and DecylTPP, a non-antioxidant control, suggesting non-specific actions of the positively-charged mitochondrial targeting moiety (67, 140, 142). Such events were associated with a compensatory elevation of glycolysis and induction of acinar cell death. The results of this study also indicated that ROS exert a significant negative feedback control of basal respiration, potentially by effects on uncoupling proteins thereby determining basal metabolic function (9, 27, 58). Increasing evidence points to important physiological signaling roles of ROS (55, 61, 71): therefore, an important consideration when addressing mitochondrial dysfunction by quenching free radicals is the potential consequences of disrupting normal physiological organellar function.
C. Inflammatory response: bioenergetics changes in blood cells
While most work elucidating bioenergetics changes related to acute pancreatitis has been carried out in pancreatic cells and tissues, changes in blood cells linked to the generation and propagation of a systemic inflammatory response are likely to be of significance (118, 151). The bioenergetics changes in circulating leukocytes are only partially understood, not least because investigations encounter significant technical difficulties during isolation, including potential damage to cells that might induce artefacts. Previously, a modest (1.5-fold) increase in basal respiration was found in total peripheral blood mononuclear cells isolated from acute pancreatitis patients, although ATP production was unaltered (34). Our recent work has shown subset-specific bioenergetics alterations occur in leukocytes from acute pancreatitis patients that imply functional alterations linked to clinical disease progression (122). Separation of blood cell subtypes from acute pancreatitis patients was feasible for detailed bioenergetics evaluations using Seahorse XF flux analysis. The study found no difference in basal respiration in blood leukocyte subtypes between acute pancreatitis patients and healthy controls. However, when the mitochondria were challenged with a “stress test” (by manipulation of the respiratory chain with rotenone and antimycin (complexes I & III), oligomycin (ATP synthase) and FCCP (protonophore)(6, 7, 28)) it was revealed that patient lymphocytes possessed decreased maximal respiration and spare respiratory capacity, indicating functional impairment. Furthermore, a diminished oxidative burst was evident in neutrophils from acute pancreatitis patients, compared to healthy controls, that may indicate compromised activity in this cell type, whereas this was enhanced in both monocytes and lymphocytes; further work is necessary to determine the bases for such altered bioenergetics in circulating blood cells and whether bioenergetics analysis might be predictive of acute pancreatitis severity and thereby assist prognosis.
D. Therapeutic aspects: maintenance of ATP provision
How may detrimental bioenergetics changes linked to development of acute pancreatitis be prevented? Recent attention has focused on inhibition of the Ca2+ overload machinery that causes mitochondrial dysfunction. For example, preclinical studies in murine pancreatic acinar cells have shown that inhibition of Orai1, expressed in pancreatic acinar cells (103), prevented sustained Ca2+ increases induced by fatty acid ethyl esters, coupled with maintenance of mitochondrial membrane potential and a reduction of necrosis (64). Prevention of the toxic effects of acute pancreatitis precipitants by Orai1 inhibitors GSK-7975A (GlaxoSmithKline) and CM4620 (CalciMedica) was further demonstrated in human pancreatic acinar cells, and the potential for acute pancreatitis treatment indicated from their efficacy in multiple in vivo experimental models (CER-AP, TLCS-AP and FAEE-AP) (175). More recently, important protective actions of the Orai1 inhibitor CM4620 on immune cells were shown that contribute to the beneficial outcomes in acute pancreatitis (172) and this agent is currently being evaluated in Phase II clinical trials (155). Interestingly, a recent study has also identified the involvement of TRPM2 channels in Ca2+ entry in pancreatic acinar cells and inhibition of these redox-sensitive channels may provide a novel approach to treat biliary acute pancreatitis (60). As mentioned above, prevention of the downstream effects of Ca2+ overload induced by acute pancreatitis precipitants on mitochondria is considered a promising therapeutic strategy, with new MPTP inhibitors in development and evaluation in preclinical models (2, 82, 124, 163, 164). Despite a lack of clarity about the composition of the MPTP, most effort has been directed towards inhibition of the modulatory protein CypD, a prime target in multiple pathologies (30). Genetic deletion of CypD or pharmacological inhibition with cyclosporin A or its analogue NIM811 treatment effectively reduced histological damage and biochemical alterations in multiple acute pancreatitis models (23, 124, 163). Furthermore, since ATP supplementation ameliorated Ca2+ clearance from the cytosol and prevented damage in acinar and ductal cells, there is potential to prevent necrotic damage in acute pancreatitis by boosting cellular energy provision (45, 84, 106). However, development of an effective means to achieve this in patients is challenging; ATP does not effectively cross cell membranes and is subject to degradation by enzymatic action. Nevertheless, studies using liposomes as a vehicle to deliver ATP have been shown to exert protective actions against damage in heart, liver and brain (96, 99, 166). Notably a recent study showed that enhancing cellular energy production with galactose ameliorated asparaginase- and FAEE-induced acute pancreatitis (134), indicating potential therapeutic value of manipulating energy levels in acute pancreatitis patients. Currently a multicentre randomized double-blind clinical (GOULASH) trial is being conducted to assess high versus low energy administration in the early phase of acute pancreatitis (116). Previously, enteral nutrition has been shown to significantly decrease mortality and frequency of multiorgan failure in severe acute pancreatitis (115, 131) and results of the GOULASH trial are awaited with interest.
VI. References
- Ahuja M, Schwartz DM, Tandon M, Son A, Zeng M, Swaim W, Eckhaus M, Hoffman V, Cui Y, Xiao B, Worley PF, and Muallem S. Orai1-Mediated Antimicrobial Secretion from Pancreatic Acini Shapes the Gut Microbiome and Regulates Gut Innate Immunity. Cell Metab 25: 635-646, 2017. PMID: 28273482.
- Antonucci S, Di Sante M, Sileikyte J, Deveraux J, Bauer T, Bround MJ, Menabò R, Paillard M, Alanova P, Carraro M, Ovize M, Molkentin JD, Cohen M, Forte MA, Bernardi P, Di Lisa F, and Murphy E. A novel class of cardioprotective small-molecule PTP inhibitors. Pharmacol Res 151: 104548, 2020. PMID: 31759087.
- Araya S, Kuster E, Gluch D, Mariotta L, Lutz C, Reding TV, Graf R, Verrey F, and Camargo SMR. Exocrine pancreas glutamate secretion help to sustain enterocyte nutritional needs under protein restriction. Am J Physiol Gastrointest Liver Physiol 314: G517-G536, 2018. PMID: 29167114.
- Armstrong JA, Cash N, Soares PM, Souza MH, Sutton R, and Criddle DN. Oxidative stress in acute pancreatitis: lost in translation? Free Radic Res 47: 917-933, 2013. PMID: 23952531.
- Armstrong JA, Cash NJ, Morton JC, Tepikin AV, Sutton R, and Criddle DN. Mitochondrial Targeting of Antioxidants Alters Pancreatic Acinar Cell Bioenergetics and Determines Cell Fate. Int J Mol Sci 20: 2019. PMID: 30959771.
- Armstrong JA, Cash NJ, Ouyang Y, Morton JC, Chvanov M, Latawiec D, Awais M, Tepikin AV, Sutton R, and Criddle DN. Oxidative stress alters mitochondrial bioenergetics and modifies pancreatic cell death independently of cyclophilin D, resulting in an apoptosis-to-necrosis shift. J Biol Chem 293: 8032-8047, 2018. PMID: 29626097.
- Armstrong JA, Sutton R, and Criddle DN. Pancreatic Acinar Cell Preparation for Oxygen Consumption and Lactate Production Analysis. Bio-protocol 10: e3627, 2020. DOI: 10.21769/BioProtoc.3627.
- Armstrong JS, Yang H, Duan W, and Whiteman M. Cytochrome bc1 regulates the mitochondrial permeability transition by two distinct pathways. J Biol Chem 279: 50420-50428, 2004. PMID: 15364912.
- Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, Couplan E, Alves-Guerra MC, Goubern M, Surwit R, Bouillaud F, Richard D, Collins S, and Ricquier D. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet 26: 435-439, 2000. PMID: 11101840.
- Asin-Cayuela J, Manas AR, James AM, Smith RA, and Murphy MP. Fine-tuning the hydrophobicity of a mitochondria-targeted antioxidant. FEBS Lett 571: 9-16, 2004. PMID: 15280009.
- Baggaley EM, Elliott AC, and Bruce JI. Oxidant-induced inhibition of the plasma membrane Ca2+-ATPase in pancreatic acinar cells: role of the mitochondria. Am J Physiol Cell Physiol 295: C1247-1260, 2008. PMID: 18787078.
- Barrow SL, Voronina SG, da Silva Xavier G, Chvanov MA, Longbottom RE, Gerasimenko OV, Petersen OH, Rutter GA, and Tepikin AV. ATP depletion inhibits Ca2+ release, influx and extrusion in pancreatic acinar cells but not pathological Ca2+ responses induced by bile. Pflugers Arch 455: 1025-1039, 2008. PMID: 17952455.
- Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, and Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem 280: 18558-18561, 2005. PMID: 15792954.
- Bauduin H, Colin M, and Dumont JE. Energy sources for protein synthesis and enzymatic secretion in rat pancreas in vitro. Biochim Biophys Acta 174: 722-733, 1969. PMID: 5776194.
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, and Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476: 341-345, 2011. PMID: 21685886.
- Bernardi P. The mitochondrial permeability transition pore: a mystery solved? Front Physiol 4: 95, 2013. PMID: 23675351.
- Bernardi P. Why F-ATP Synthase Remains a Strong Candidate as the Mitochondrial Permeability Transition Pore. Front Physiol 9: 1543, 2018. PMID: 30443222.
- Betzenhauser MJ, Wagner LE, 2nd, Iwai M, Michikawa T, Mikoshiba K, and Yule DI. ATP modulation of Ca2+ release by type-2 and type-3 inositol (1, 4, 5)-triphosphate receptors. Differing ATP sensitivities and molecular determinants of action. J Biol Chem 283: 21579-21587, 2008. PMID: 18505727.
- Bhatia M, Wallig MA, Hofbauer B, Lee HS, Frossard JL, Steer ML, and Saluja AK. Induction of apoptosis in pancreatic acinar cells reduces the severity of acute pancreatitis. Biochem Biophys Res Comm 246: 476-483, 1998. PMID: 9610387.
- Bhosale G, Sharpe JA, Koh A, Kouli A, Szabadkai G, and Duchen MR. Pathological consequences of MICU1 mutations on mitochondrial calcium signalling and bioenergetics. Biochim Biophys Acta Mol Cell Res 1864: 1009-1017, 2017. PMID: 28132899.
- Bhosale G, Sharpe JA, Sundier SY, and Duchen MR. Calcium signaling as a mediator of cell energy demand and a trigger to cell death. Ann N Y Acad Sci 1350: 107-116, 2015. PMID: 26375864.
- Biczo G, Hegyi P, Dosa S, Shalbuyeva N, Berczi S, Sinervirta R, Hracsko Z, Siska A, Kukor Z, Jarmay K, Venglovecz V, Varga IS, Ivanyi B, Alhonen L, Wittmann T, Gukovskaya A, Takacs T, and Rakonczay Z, Jr. The crucial role of early mitochondrial injury in L-lysine-induced acute pancreatitis. Antioxid Redox Signal 15: 2669-2681, 2011. PMID: 21644850.
- Biczo G, Vegh ET, Shalbueva N, Mareninova OA, Elperin J, Lotshaw E, Gretler S, Lugea A, Malla SR, Dawson D, Ruchala P, Whitelegge J, French SW, Wen L, Husain SZ, Gorelick FS, Hegyi P, Rakonczay Z, Jr., Gukovsky I, and Gukovskaya AS. Mitochondrial Dysfunction, Through Impaired Autophagy, Leads to Endoplasmic Reticulum Stress, Deregulated Lipid Metabolism, and Pancreatitis in Animal Models. Gastroenterology 154: 689-703, 2018. PMID: 29074451.
- Bolender RP. Stereological analysis of the guinea pig pancreas. I. Analytical model and quantitative description of nonstimulated pancreatic exocrine cells. J Cell Biol 61: 269-287, 1974. PMID: 4363955.
- Bondarenko AI, Jean-Quartier C, Parichatikanond W, Alam MR, Waldeck-Weiermair M, Malli R, and Graier WF. Mitochondrial Ca(2+) uniporter (MCU)-dependent and MCU-independent Ca(2+) channels coexist in the inner mitochondrial membrane. Pflugers Arch 466: 1411-1420, 2014. PMID: 24162235.
- Booth DM, Murphy JA, Mukherjee R, Awais M, Neoptolemos JP, Gerasimenko OV, Tepikin AV, Petersen OH, Sutton R, and Criddle DN. Reactive oxygen species induced by bile acid induce apoptosis and protect against necrosis in pancreatic acinar cells. Gastroenterology 140: 2116-2125, 2011. PMID: 21354148.
- Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakay JL, and Parker N. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med 37: 755-767, 2004. PMID: 15304252.
- Brand MD, and Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J 435: 297-312, 2011. PMID: 21726199.
- Brini M, and Carafoli E. Calcium pumps in health and disease. Physiol Rev 89: 1341-1378, 2009. PMID: 19789383.
- Briston T, Selwood DL, Szabadkai G, and Duchen MR. Mitochondrial Permeability Transition: A Molecular Lesion with Multiple Drug Targets. Trends Pharmacol Sci 40: 50-70, 2019. PMID: 30527591.
- Bround MJ, Bers DM, and Molkentin JD. A 20/20 view of ANT function in mitochondrial biology and necrotic cell death. J Mol Cell Cardiol 144: A3-A13, 2020. PMID: 32454061.
- Campanella M, Casswell E, Chong S, Farah Z, Wieckowski MR, Abramov AY, Tinker A, and Duchen MR. Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1. Cell Metab 8: 13-25, 2008. PMID: 18590689.
- Chacko BK, Reily C, Srivastava A, Johnson MS, Ye Y, Ulasova E, Agarwal A, Zinn KR, Murphy MP, Kalyanaraman B, and Darley-Usmar V. Prevention of diabetic nephropathy in Ins2+/-AkitaJ mice by the mitochondria-targeted therapy MitoQ. Biochem J 432: 9-19, 2010. PMID: 20825366.
- Chakraborty M, Hickey AJ, Petrov MS, Macdonald JR, Thompson N, Newby L, Sim D, Windsor JA, and Phillips AR. Mitochondrial dysfunction in peripheral blood mononuclear cells in early experimental and clinical acute pancreatitis. Pancreatology 16: 739-747, 2016. PMID: 27473495.
- Chandra R, and Liddle RA. Regulation of Pancreatic Secretion. Pancreapedia: Exocrine Pancreas Knowledge Base 2020. DOI: 10.3998/panc.2020.14.
- Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S, James AM, Cocheme HM, Reinhold J, Lilley KS, Partridge L, Fearnley IM, Robinson AJ, Hartley RC, Smith RA, Krieg T, Brookes PS, and Murphy MP. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat Med 19: 753-759, 2013. PMID: 23708290.
- Chvanov M, De Faveri F, Moore D, Sherwood MW, Awais M, Voronina S, Sutton R, Criddle DN, Haynes L, and Tepikin AV. Intracellular rupture, exocytosis and actin interaction of endocytic vacuoles in pancreatic acinar cells: initiating events in acute pancreatitis. J Physiol 596: 2547-2564, 2018. PMID: 29717784.
- Chvanov M, Huang W, Jin T, Wen L, Armstrong J, Elliot V, Alston B, Burdyga A, Criddle DN, Sutton R, and Tepikin AV. Novel lipophilic probe for detecting near-membrane reactive oxygen species responses and its application for studies of pancreatic acinar cells: effects of pyocyanin and L-ornithine. Antioxid Redox Signal 22: 451-464, 2015. PMID: 24635199.
- Chvanov M, Voronina S, Zhang X, Telnova S, Chard R, Ouyang Y, Armstrong J, Tanton H, Awais M, Latawiec D, Sutton R, Criddle DN, and Tepikin AV. Knockout of the Mitochondrial Calcium Uniporter Strongly Suppresses Stimulus-Metabolism Coupling in Pancreatic Acinar Cells but Does Not Reduce Severity of Experimental Acute Pancreatitis. Cells 9: 2020. PMID: 32516955.
- Criddle DN. Keeping mitochondria happy - benefits of a pore choice in acute pancreatitis. J Physiol 597: 5741-5742, 2019. PMID: 31670384.
- Criddle DN. Reactive oxygen species, Ca(2+) stores and acute pancreatitis; a step closer to therapy? Cell Calcium 60: 180-189, 2016. PMID: 27229361.
- Criddle DN. The role of fat and alcohol in acute pancreatitis: A dangerous liaison. Pancreatology 15: S6-S12, 2015. PMID: 25845855.
- Criddle DN, Gerasimenko JV, Baumgartner HK, Jaffar M, Voronina S, Sutton R, Petersen OH, and Gerasimenko OV. Calcium signalling and pancreatic cell death: apoptosis or necrosis? Cell Death Differ 14: 1285-1294, 2007. PMID: 17431416.
- Criddle DN, Gillies S, Baumgartner-Wilson HK, Jaffar M, Chinje EC, Passmore S, Chvanov M, Barrow S, Gerasimenko OV, Tepikin AV, Sutton R, and Petersen OH. Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J Biol Chem 281: 40485-40492, 2006. PMID: 17088248.
- Criddle DN, Murphy J, Fistetto G, Barrow S, Tepikin AV, Neoptolemos JP, Sutton R, and Petersen OH. Fatty Acid Ethyl Esters Cause Pancreatic Calcium Toxicity via Inositol Trisphosphate Receptors and Loss of ATP Synthesis. Gastroenterology 130: 781-793, 2006. PMID: 16530519.
- Criddle DN, Raraty MG, Neoptolemos JP, Tepikin AV, Petersen OH, and Sutton R. Ethanol toxicity in pancreatic acinar cells: mediation by nonoxidative fatty acid metabolites. Proc Natl Acad Sci USA 101: 10738-10743, 2004. PMID: 15247419.
- Csordas G, Golenar T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, Moffat C, Weaver D, Perez SF, Bogorad R, Koteliansky V, Adijanto J, Mootha VK, and Hajnoczky G. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab 17: 976-987, 2013. PMID: 23747253.
- Cuthbertson CM, and Christophi C. Disturbances of the microcirculation in acute pancreatitis. Br J Surg 93: 518-530, 2006. PMID: 16607683.
- Dare AJ, Bolton EA, Pettigrew GJ, Bradley JA, Saeb-Parsy K, and Murphy MP. Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Biol 5: 163-168, 2015. PMID: 25965144.
- Dashdorj A, Jyothi KR, Lim S, Jo A, Nguyen MN, Ha J, Yoon KS, Kim HJ, Park JH, Murphy MP, and Kim SS. Mitochondria-targeted antioxidant MitoQ ameliorates experimental mouse colitis by suppressing NLRP3 inflammasome-mediated inflammatory cytokines. BMC Med 11: 178, 2013. PMID: 23915129.
- De Stefani D, Raffaello A, Teardo E, Szabo I, and Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476: 336-340, 2011. PMID: 21685888.
- Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta 1787: 1309-1316, 2009. PMID: 19413950.
- Diczfalusy MA, Bjorkhem I, Einarsson C, Hillebrant CG, and Alexson SE. Characterization of enzymes involved in formation of ethyl esters of long-chain fatty acids in humans. J Lipid Res 42: 1025-1032, 2001. PMID: 11441128.
- Dolman NJ, Gerasimenko JV, Gerasimenko OV, Voronina SG, Petersen OH, and Tepikin AV. Stable Golgi-mitochondria complexes and formation of Golgi Ca2+ gradients in pancreatic acinar cells. J Biol Chem 280: 15794-15799, 2005. PMID: 15722348.
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev 82: 47-95, 2002. PMID: 11773609.
- Duchen MR. Ca2+-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochem J 283 ( Pt 1): 41-50, 1992. PMID: 1373604.
- Duchen MR, Surin A, and Jacobson J. Imaging mitochondrial function in intact cells. Methods Enzymol 361: 353-389, 2003. PMID: 12624920.
- Echtay KS, Murphy MP, Smith RA, Talbot DA, and Brand MD. Superoxide activates mitochondrial uncoupling protein 2 from the matrix side. Studies using targeted antioxidants. J Biol Chem 277: 47129-47135, 2002. PMID: 12372827.
- Elrod JW, and Molkentin JD. Physiologic Functions of Cyclophilin D and the Mitochondrial Permeability Transition Pore. Circ J 77: 1111-1122, 2013. PMID: 23538482.
- Fanczal J, Pallagi P, Gorog M, Diszhazi G, Almassy J, Madacsy T, Varga A, Csernay-Biro P, Katona X, Toth E, Molnar R, Rakonczay Z, Jr., Hegyi P, and Maleth J. TRPM2-mediated extracellular Ca2+ entry promotes acinar cell necrosis in biliary acute pancreatitis. J Physiol 598: 1253-1270, 2020. PMID: 31917868.
- Finkel T. Signal transduction by reactive oxygen species. J Cell Biol 194: 7-15, 2011. PMID: 21746850.
- Futatsugi A, Nakamura T, Yamada MK, Ebisui E, Nakamura K, Uchida K, Kitaguchi T, Takahashi-Iwanaga H, Noda T, Aruga J, and Mikoshiba K. IP3 receptor types 2 and 3 mediate exocrine secretion underlying energy metabolism. Science 309: 2232-2234, 2005. PMID: 16195467.
- Gamberucci A, Innocenti B, Fulceri R, Banhegyi G, Giunti R, Pozzan T, and Benedetti A. Modulation of Ca2+ influx dependent on store depletion by intracellular adenine-guanine nucleotide levels. J Biol Chem 269: 23597-23602, 1994. PMID: 8089128.
- Gerasimenko JV, Gryshchenko O, Ferdek PE, Stapleton E, Hebert TO, Bychkova S, Peng S, Begg M, Gerasimenko OV, and Petersen OH. Ca2+ release-activated Ca2+ channel blockade as a potential tool in antipancreatitis therapy. Proc Natl Acad Sci USA 110: 13186-13191, 2013. PMID: 23878235.
- Giorgio V, Guo L, Bassot C, Petronilli V, and Bernardi P. Calcium and regulation of the mitochondrial permeability transition. Cell Calcium 70: 56-63, 2018. PMID: 28522037.
- Glitsch MD, Bakowski D, and Parekh AB. Store-operated Ca2+ entry depends on mitochondrial Ca2+ uptake. EMBO J 21: 6744-6754, 2002. PMID: 12485995.
- Gottwald EM, Duss M, Bugarski M, Haenni D, Schuh CD, Landau EM, and Hall AM. The targeted anti-oxidant MitoQ causes mitochondrial swelling and depolarization in kidney tissue. Physiol Rep 6: e13667, 2018. PMID: 29611340.
- Grover GJ, Marone PA, Koetzner L, and Seto-Young D. Energetic signalling in the control of mitochondrial F1F0 ATP synthase activity in health and disease. Int J Biochem Cell Biol 40: 2698-2701, 2008. PMID: 18707016.
- Hajnoczky G, Robb-Gaspers LD, Seitz MB, and Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell 82: 415-424, 1995. PMID: 7634331.
- Halangk W, Matthias R, Schild L, Meyer F, Schulz HU, and Lippert H. Effect of supramaximal cerulein stimulation on mitochondrial energy metabolism in rat pancreas. Pancreas 16: 88-95, 1998. PMID: 9436868.
- Hamanaka RB, and Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci 35: 505-513, 2010. PMID: 20430626.
- Hamilton J, Brustovetsky T, Rysted JE, Lin Z, Usachev YM, and Brustovetsky N. Deletion of mitochondrial calcium uniporter incompletely inhibits calcium uptake and induction of the permeability transition pore in brain mitochondria. J Biol Chem 293: 15652-15663, 2018. PMID: 30154242.
- He J, Carroll J, Ding S, Fearnley IM, and Walker JE. Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase. Proc Natl Acad Sci USA 114: 9086-9091, 2017. PMID: 28784775.
- He J, Ford HC, Carroll J, Ding S, Fearnley IM, and Walker JE. Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc Natl Acad Sci USA 114: 3409-3414, 2017. PMID: 28289229.
- He L, and Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS letters 512: 1-7, 2002. PMID: 11852041.
- Hegyi P, and Petersen OH. The exocrine pancreas: the acinar-ductal tango in physiology and pathophysiology. Rev Physiol Biochem Pharmacol 165: 1-30, 2013. PMID: 23881310.
- Hirano T, Manabe T, and Tobe T. Protective effects of gabexate mesilate (FOY) against impaired pancreatic energy metabolism in rat acute pancreatitis induced by caerulein. Life Sci 49: PL179-184, 1991. PMID: 1719326.
- Hofer AM, Curci S, Machen TE, and Schulz I. ATP regulates calcium leak from agonist-sensitive internal calcium stores. FASEB J 10: 302-308, 1996. PMID: 8641563.
- Hoth M, Fanger CM, and Lewis RS. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. J Cell Biol 137: 633-648, 1997. PMID: 9151670.
- Huang W, Booth DM, Cane MC, Chvanov M, Javed MA, Elliott VL, Armstrong JA, Dingsdale H, Cash N, Li Y, Greenhalf W, Mukherjee R, Kaphalia BS, Jaffar M, Petersen OH, Tepikin AV, Sutton R, and Criddle DN. Fatty acid ethyl ester synthase inhibition ameliorates ethanol-induced Ca2+-dependent mitochondrial dysfunction and acute pancreatitis. Gut 63: 1313-1324, 2014. PMID: 24162590.
- Huang W, Cash N, Wen L, Szatmary P, Mukherjee R, Armstrong J, Chvanov M, Tepikin AV, Murphy MP, Sutton R, and Criddle DN. Effects of the mitochondria-targeted antioxidant mitoquinone in murine acute pancreatitis. Mediators Inflamm 2015: 901780, 2015. PMID: 25878403.
- Javed MA, Wen L, Awais M, Latawiec D, Huang W, Chvanov M, Schaller S, Bordet T, Michaud M, Pruss R, Tepikin A, Criddle D, and Sutton R. TRO40303 Ameliorates Alcohol-Induced Pancreatitis Through Reduction of Fatty Acid Ethyl Ester-Induced Mitochondrial Injury and Necrotic Cell Death. Pancreas 47: 18-24, 2018. PMID: 29200128.
- Johnson PR, Dolman NJ, Pope M, Vaillant C, Petersen OH, Tepikin AV, and Erdemli G. Non-uniform distribution of mitochondria in pancreatic acinar cells. Cell Tissue Res 313: 37-45, 2003. PMID: 12838407.
- Judak L, Hegyi P, Rakonczay Z, Jr., Maleth J, Gray MA, and Venglovecz V. Ethanol and its non-oxidative metabolites profoundly inhibit CFTR function in pancreatic epithelial cells which is prevented by ATP supplementation. Pflugers Arch 466: 549-562, 2014. PMID: 23948742.
- Kaiser AM, Saluja AK, Sengupta A, Saluja M, and Steer ML. Relationship between severity, necrosis, and apoptosis in five models of experimental acute pancreatitis. Am J Physiol 269: C1295-C1304, 1995. PMID: 7491921.
- Kamer KJ, and Mootha VK. MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep 15: 299-307, 2014. PMID: 24503055.
- Kanno T, Saito A, and Ikei N. Dose-dependent effect of acetylcholine stimulating respiratory chain and secretion of isolated perfused rat pancreas. Biomed Res 4: 175-186, 1983. DOI: 10.2220/biomedres.4.175.
- Karch J, Bround MJ, Khalil H, Sargent MA, Latchman N, Terada N, Peixoto PM, and Molkentin JD. Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci Adv 5: eaaw4597, 2019. PMID: 31489369.
- Karch J, and Molkentin JD. Identity of the elusive mitochondrial permeability transition pore: what it might be, what it was, and what it still could be. Curr Opin Physiol 3: 57-62, 2018.
- Kasai H, Li YX, and Miyashita Y. Subcellular distribution of Ca2+ release channels underlying Ca2+ waves and oscillations in exocrine pancreas. Cell 74: 669-677, 1993. PMID: 8395348.
- Katona M, Hegyi P, Kui B, Balla Z, Rakonczay Z, Jr., Razga Z, Tiszlavicz L, Maleth J, and Venglovecz V. A novel, protective role of ursodeoxycholate in bile-induced pancreatic ductal injury. Am J Physiol Gastrointest Liver Physiol 310: G193-204, 2016. PMID: 26608189.
- Kim MS, Hong JH, Li Q, Shin DM, Abramowitz J, Birnbaumer L, and Muallem S. Deletion of TRPC3 in mice reduces store-operated Ca2+ influx and the severity of acute pancreatitis. Gastroenterology 137: 1509-1517, 2009. PMID: 19622358.
- Kinnala PJ, Kuttila KT, Gronroos JM, Havia TV, Nevalainen TJ, and Niinikoski JH. Pancreatic tissue perfusion in experimental acute pancreatitis. Eur J Surg 167: 689-694, 2001. PMID: 11759740.
- Kloppel G, and Maillet B. Pathology of acute and chronic pancreatitis. Pancreas 8: 659-670, 1993. PMID: 8255882.
- Knoefel WT, Kollias N, Warshaw AL, Waldner H, Nishioka NS, and Rattner DW. Pancreatic microcirculatory changes in experimental pancreatitis of graded severity in the rat. Surgery 116: 904-913, 1994. PMID: 7940196.
- Konno H, Matin AF, Maruo Y, Nakamura S, and Baba S. Liposomal ATP protects the liver from injury during shock. Eur Surg Res 28: 140-145, 1996. PMID: 8834372.
- Korc M, Williams JA, and Goldfine ID. Stimulation of the glucose transport system in isolated mouse pancreatic acini by cholecystokinin and analogues. J Biol Chem 254: 7624-7629, 1979. PMID: 381288.
- Kosowski H, Schild L, Kunz D, and Halangk W. Energy metabolism in rat pancreatic acinar cells during anoxia and reoxygenation. Biochim Biophys Acta 1367: 118-126, 1998. PMID: 9784620.
- Laham A, Claperon N, Durussel JJ, Fattal E, Delattre J, Puisieux F, Couvreur P, and Rossignol P. Intracarotidal administration of liposomally-entrapped ATP: improved efficiency against experimental brain ischemia. Pharmacol Res Commun 20: 699-705, 1988. PMID: 3212008.
- Laukkarinen JM, Van Acker GJ, Weiss ER, Steer ML, and Perides G. A mouse model of acute biliary pancreatitis induced by retrograde pancreatic duct infusion of Na-taurocholate. Gut 56: 1590-1598, 2007. PMID: 17591621.
- Lehninger AL, and Wadkins CL. Oxidative phosphorylation. Annu Rev Biochem 31: 47-78, 1962. PMID: 14463786.
- Lowes DA, Webster NR, Murphy MP, and Galley HF. Antioxidants that protect mitochondria reduce interleukin-6 and oxidative stress, improve mitochondrial function, and reduce biochemical markers of organ dysfunction in a rat model of acute sepsis. Br J Anaesth 110: 472-480, 2013. PMID: 23381720.
- Lur G, Haynes LP, Prior IA, Gerasimenko OV, Feske S, Petersen OH, Burgoyne RD, and Tepikin AV. Ribosome-free terminals of rough ER allow formation of STIM1 puncta and segregation of STIM1 from IP3 receptors. Curr Biol 19: 1648-1653, 2009. PMID: 19765991.
- Lüthen RE, Niederau C, Ferrell LD, and Grendell JH. Energy metabolism in mouse pancreas in response to different dosages of a CCK analogue. Pancreas 11: 141-146, 1995. PMID: 7479670.
- Maleth J, Balazs A, Pallagi P, Balla Z, Kui B, Katona M, Judak L, Nemeth I, Kemeny LV, Rakonczay Z, Jr., Venglovecz V, Foldesi I, Peto Z, Somoracz A, Borka K, Perdomo D, Lukacs GL, Gray MA, Monterisi S, Zaccolo M, Sendler M, Mayerle J, Kuhn JP, Lerch MM, Sahin-Toth M, and Hegyi P. Alcohol disrupts levels and function of the cystic fibrosis transmembrane conductance regulator to promote development of pancreatitis. Gastroenterology 148: 427-439 e416, 2015. PMID: 25447846.
- Maleth J, Hegyi P, Rakonczay Z, Jr., and Venglovecz V. Breakdown of bioenergetics evoked by mitochondrial damage in acute pancreatitis: Mechanisms and consequences. Pancreatology 15: S18-22, 2015. PMID: 26162756.
- Maleth J, Venglovecz V, Razga Z, Tiszlavicz L, Rakonczay Z, and Hegyi P. Non-conjugated chenodeoxycholate induces severe mitochondrial damage and inhibits bicarbonate transport in pancreatic duct cells. Gut 60: 136-138, 2011. PMID: 20732916.
- Malla SR, Krueger B, Wartmann T, Sendler M, Mahajan UM, Weiss FU, Thiel FG, De Boni C, Gorelick FS, Halangk W, Aghdassi AA, Reinheckel T, Gukovskaya AS, Lerch MM, and Mayerle J. Early trypsin activation develops independently of autophagy in caerulein-induced pancreatitis in mice. Cell Mol Life Sci 77: 1811-1825, 2020. PMID: 31363815.
- Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ, Rothberg BS, Mak DO, Foskett JK, and Madesh M. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell 151: 630-644, 2012. PMID: 23101630.
- Mammucari C, Raffaello A, Vecellio Reane D, Gherardi G, De Mario A, and Rizzuto R. Mitochondrial calcium uptake in organ physiology: from molecular mechanism to animal models. Pflugers Arch 470: 1165-1179, 2018. PMID: 29541860.
- Mankad P, James A, Siriwardena AK, Elliott AC, and Bruce JI. Insulin protects pancreatic acinar cells from cytosolic calcium overload and inhibition of plasma membrane calcium pump. J Biol Chem 287: 1823-1836, 2012. PMID: 22128146.
- Manko BO, and Manko VV. Mechanisms of respiration intensification of rat pancreatic acini upon carbachol-induced Ca2+ release. Acta Physiol (Oxf) 208: 387-399, 2013. PMID: 23692873.
- Mareninova OA, Sung KF, Hong P, Lugea A, Pandol SJ, Gukovsky I, and Gukovskaya AS. Cell death in pancreatitis: caspases protect from necrotizing pancreatitis. J Biol Chem 281: 3370-3381, 2006. PMID: 16339139.
- Marriott I, and Mason MJ. ATP depletion inhibits capacitative Ca2+ entry in rat thymic lymphocytes. Am J Physiol 269: C766-774, 1995. PMID: 7573408.
- Marta K, Farkas N, Szabo I, Illes A, Vincze A, Par G, Sarlos P, Bajor J, Szucs A, Czimmer J, Mosztbacher D, Parniczky A, Szemes K, Pecsi D, and Hegyi P. Meta-Analysis of Early Nutrition: The Benefits of Enteral Feeding Compared to a Nil Per Os Diet Not Only in Severe, but Also in Mild and Moderate Acute Pancreatitis. Int J Mol Sci 17: 2016. PMID: 27775609.
- Marta K, Szabo AN, Pecsi D, Varju P, Bajor J, Godi S, Sarlos P, Miko A, Szemes K, Papp M, Tornai T, Vincze A, Marton Z, Vincze PA, Lanko E, Szentesi A, Molnar T, Hagendorn R, Faluhelyi N, Battyani I, Kelemen D, Papp R, Miseta A, Verzar Z, Lerch MM, Neoptolemos JP, Sahin-Toth M, Petersen OH, Hegyi P, and Hung Panc St Gr. High versus low energy administration in the early phase of acute pancreatitis (GOULASH trial): protocol of a multicentre randomised double-blind clinical trial. BMJ Open 7: e015874, 2017. PMID: 28912191.
- Matsumoto T, Kanno T, Seo Y, Murakami M, and Watari H. Dose effects of cholecystokinin and acetylcholine on phosphorus compounds and secretory responses in isolated perfused pancreas of rat. Jpn J Physiol 41: 483-492, 1991. PMID: 1960892.
- Mayerle J, Sendler M, Hegyi E, Beyer G, Lerch MM, and Sahin-Toth M. Genetics, Cell Biology, and Pathophysiology of Pancreatitis. Gastroenterology 156: 1951-1968 e1951, 2019. PMID: 30660731.
- McCormack JG, Halestrap AP, and Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev 70: 391-425, 1990. PMID: 2157230.
- McEntee G, Leahy A, Cottell D, Dervan P, McGeeney K, and Fitzpatrick JM. Three-dimensional morphological study of the pancreatic microvasculature in caerulein-induced experimental pancreatitis. Br J Surg 76: 853-855, 1989. PMID: 2765844.
- Morton JC, Armstrong J, Cash N, Ouyang Y, Tepikin A, Sutton R, and Criddle DN. Pathophysiological Modulation Of Pancreatic Acinar Cell Bioenergetics By Cholecystokinin. Pancreas 45: 1527 (Abstract), 2016.
- Morton JC, Armstrong JA, Sud A, Tepikin AV, Sutton R, and Criddle DN. Altered Bioenergetics of Blood Cell Sub-Populations in Acute Pancreatitis Patients. J Clin Med 8: 2019. PMID: 31847184.
- Muallem S, Beeker T, and Pandol SJ. Role of Na+/Ca2+ exchange and the plasma membrane Ca2+ pump in hormone-mediated Ca2+ efflux from pancreatic acini. J Membr Biol 102: 153-162, 1988. PMID: 2458473.
- Mukherjee R, Mareninova OA, Odinokova IV, Huang W, Murphy J, Chvanov M, Javed MA, Wen L, Booth DM, Cane MC, Awais M, Gavillet B, Pruss RM, Schaller S, Molkentin JD, Tepikin AV, Petersen OH, Pandol SJ, Gukovsky I, Criddle DN, Gukovskaya AS, Sutton R, and NIH Panc Biomed Res Unit. Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: inhibition prevents acute pancreatitis by protecting production of ATP. Gut 65: 1333-1346, 2016. PMID: 26071131.
- Murphy JA, Criddle DN, Sherwood M, Chvanov M, Mukherjee R, McLaughlin E, Booth D, Gerasimenko JV, Raraty MG, Ghaneh P, Neoptolemos JP, Gerasimenko OV, Tepikin AV, Green GM, Reeve JR, Jr., Petersen OH, and Sutton R. Direct activation of cytosolic Ca2+ signaling and enzyme secretion by cholecystokinin in human pancreatic acinar cells. Gastroenterology 135: 632-641, 2008. PMID: 18555802.
- Murphy MP, and Hartley RC. Mitochondria as a therapeutic target for common pathologies. Nat Rev Drug Discov 2018. PMID: 30393373.
- Nordback IH, Clemens JA, Chacko VP, Olson JL, and Cameron JL. Changes in high-energy phosphate metabolism and cell morphology in four models of acute experimental pancreatitis. Ann Surg 213: 341-349, 1991. PMID: 2009016.
- Pallagi P, Hegyi P, and Rakonczay Z, Jr. The Physiology and Pathophysiology of Pancreatic Ductal Secretion: The Background for Clinicians. Pancreas 44: 1211-1233, 2015. PMID: 26465950.
- Park HS, Betzenhauser MJ, Won JH, Chen J, and Yule DI. The type 2 inositol (1,4,5)-trisphosphate (InsP3) receptor determines the sensitivity of InsP3-induced Ca2+ release to ATP in pancreatic acinar cells. J Biol Chem 283: 26081-26088, 2008. PMID: 18658132.
- Park MK, Ashby MC, Erdemli G, Petersen OH, and Tepikin AV. Perinuclear, perigranular and sub-plasmalemmal mitochondria have distinct functions in the regulation of cellular calcium transport. EMBO J 20: 1863-1874, 2001. PMID: 11296220.
- Parniczky A, Kui B, Szentesi A, Balazs A, Szucs A, Mosztbacher D, Czimmer J, Sarlos P, Bajor J, Godi S, Vincze A, Illes A, Szabo I, Par G, Takacs T, Czako L, Szepes Z, Rakonczay Z, Izbeki F, Gervain J, Halasz A, Novak J, Crai S, Hritz I, Gog C, Sumegi J, Golovics P, Varga M, Bod B, Hamvas J, Varga-Muller M, Papp Z, Sahin-Toth M, Hegyi P, and Hung Panc St Gr. Prospective, Multicentre, Nationwide Clinical Data from 600 Cases of Acute Pancreatitis. PLoS One 11: e0165309, 2016. PMID: 27798670.
- Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabo I, De Stefani D, and Rizzuto R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell 53: 726-737, 2014. PMID: 24560927.
- Peng S, Gerasimenko JV, Tsugorka T, Gryshchenko O, Samarasinghe S, Petersen OH, and Gerasimenko OV. Calcium and adenosine triphosphate control of cellular pathology: asparaginase-induced pancreatitis elicited via protease-activated receptor 2. Philos Trans R Soc Lond B Biol Sci 371: 2016. PMID: 27377732.
- Peng S, Gerasimenko JV, Tsugorka TM, Gryshchenko O, Samarasinghe S, Petersen OH, and Gerasimenko OV. Galactose protects against cell damage in mouse models of acute pancreatitis. J Clin Invest 128: 3769-3778, 2018. PMID: 29893744.
- Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, and Mootha VK. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 467: 291-296, 2010. PMID: 20693986.
- Petersen OH, Sutton R, and Criddle DN. Failure of calcium microdomain generation and pathological consequences. Cell Calcium 40: 593-600, 2006. PMID: 17049597.
- Petersen OH, and Tepikin AV. Polarized calcium signaling in exocrine gland cells. Annu Rev Physiol 70: 273-299, 2008. PMID: 17850212.
- Pham T, MacRae CL, Broome SC, D’souza RF, Narang R, Wang HW, Mori TA, Hickey AJR, Mitchell CJ, and Merry TL. MitoQ and CoQ10 supplementation mildly suppresses skeletal muscle mitochondrial hydrogen peroxide levels without impacting mitochondrial function in middle-aged men. Eur J Appl Physiol 120: 1657-1669, 2020. PMID: 32458156.
- Plusczyk T, Westermann S, Rathgeb D, and Feifel G. Acute pancreatitis in rats: effects of sodium taurocholate, CCK-8, and Sec on pancreatic microcirculation. Am J Physiol 272: G310-320, 1997. PMID: 9124355.
- Pokrzywinski KL, Biel TG, Kryndushkin D, and Rao VA. Therapeutic Targeting of the Mitochondria Initiates Excessive Superoxide Production and Mitochondrial Depolarization Causing Decreased mtDNA Integrity. PLoS One 11: e0168283, 2016. PMID: 28030582.
- Raraty M, Ward J, Erdemli G, Vaillant C, Neoptolemos JP, Sutton R, and Petersen OH. Calcium-dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc Natl Acad Sci USA 97: 13126-13131, 2000. PMID: 11087863.
- Reily C, Mitchell T, Chacko BK, Benavides G, Murphy MP, and Darley-Usmar V. Mitochondrially targeted compounds and their impact on cellular bioenergetics. Redox Biol 1: 86-93, 2013. PMID: 23667828.
- Romac JM, Shahid RA, Swain SM, Vigna SR, and Liddle RA. Piezo1 is a mechanically activated ion channel and mediates pressure induced pancreatitis. Nat Commun 9: 1715, 2018. PMID: 29712913.
- Rooman I, Lutz C, Pinho AV, Huggel K, Reding T, Lahoutte T, Verrey F, Graf R, and Camargo SM. Amino acid transporters expression in acinar cells is changed during acute pancreatitis. Pancreatology 13: 475-485, 2013. PMID: 24075511.
- Samad A, James A, Wong J, Mankad P, Whitehouse J, Patel W, Alves-Simoes M, Siriwardena AK, and Bruce JI. Insulin protects pancreatic acinar cells from palmitoleic acid-induced cellular injury. J Biol Chem 289: 23582-23595, 2014. PMID: 24993827.
- Samanta K, Douglas S, and Parekh AB. Mitochondrial calcium uniporter MCU supports cytoplasmic Ca2+ oscillations, store-operated Ca2+ entry and Ca2+-dependent gene expression in response to receptor stimulation. PLoS One 9: e101188, 2014. PMID: 25004162.
- Samanta K, Mirams GR, and Parekh AB. Sequential forward and reverse transport of the Na+ Ca2+ exchanger generates Ca2+ oscillations within mitochondria. Nat Commun 9: 156, 2018. PMID: 29323106.
- Sanfey H, and Cameron JL. Increased capillary permeability: an early lesion in acute pancreatitis. Surgery 96: 485-491, 1984. PMID: 6206583.
- Sansbury BE, Jones SP, Riggs DW, Darley-Usmar VM, and Hill BG. Bioenergetic function in cardiovascular cells: the importance of the reserve capacity and its biological regulation. Chem Biol Interact 191: 288-295, 2011. PMID: 21147079.
- Schild L, Matthias R, Stanarius A, Wolf G, Augustin W, and Halangk W. Induction of permeability transition in pancreatic mitochondria by cerulein in rats. Mol Cell Biochem 195: 191-197, 1999. PMID: 10395083.
- Sendler M, Weiss FU, Golchert J, Homuth G, van den Brandt C, Mahajan UM, Partecke LI, Doring P, Gukovsky I, Gukovskaya AS, Wagh PR, Lerch MM, and Mayerle J. Cathepsin B-Mediated Activation of Trypsinogen in Endocytosing Macrophages Increases Severity of Pancreatitis in Mice. Gastroenterology 154: 704-718 e710, 2018. PMID: 29079517.
- Shalbueva N, Mareninova OA, Gerloff A, Yuan J, Waldron RT, Pandol SJ, and Gukovskaya AS. Effects of Oxidative Alcohol Metabolism on the Mitochondrial Permeability Transition Pore and Necrosis in a Mouse Model of Alcoholic Pancreatitis. Gastroenterology 144: 437-446 e6, 2013. PMID: 23103769.
- Šileikytė J, and Forte M. The Mitochondrial Permeability Transition in Mitochondrial Disorders. Oxid Med Cell Longev 2019: 1-11, 2019. PMID: 31191798.
- Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O'Sullivan JD, Fung V, Smith RA, Murphy MP, Taylor KM, and Protect Study G. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson's disease. Mov Disord 25: 1670-1674, 2010. PMID: 20568096.
- Stauderman KA. CRAC channels as targets for drug discovery and development. Cell Calcium 74: 147-159, 2018. PMID: 30075400.
- Straub SV, Giovannucci DR, and Yule DI. Calcium wave propagation in pancreatic acinar cells: functional interaction of inositol 1,4,5-trisphosphate receptors, ryanodine receptors, and mitochondria. J Gen Physiol 116: 547-560, 2000. PMID: 11004204.
- Swain SM, Romac JM, Shahid RA, Pandol SJ, Liedtke W, Vigna SR, and Liddle RA. TRPV4 channel opening mediates pressure-induced pancreatitis initiated by Piezo1 activation. J Clin Invest 130: 2527-2541, 2020. PMID: 31999644.
- Tanton H, Voronina S, Evans A, Armstrong J, Sutton R, Criddle DN, Haynes L, Schmid MC, Campbell F, Costello E, and Tepikin AV. F1F0-ATP Synthase Inhibitory Factor 1 in the Normal Pancreas and in Pancreatic Ductal Adenocarcinoma: Effects on Bioenergetics, Invasion and Proliferation. Front Physiol 9: 833, 2018. PMID: 30050450.
- Tepikin AV. Mitochondrial junctions with cellular organelles: Ca2+ signalling perspective. Pflugers Arch 470: 1181-1192, 2018. PMID: 29982949.
- Thorn P, Lawrie AM, Smith PM, Gallacher DV, and Petersen OH. Local and global cytosolic Ca2+ oscillations in exocrine cells evoked by agonists and inositol trisphosphate. Cell 74: 661-668, 1993. PMID: 8395347.
- Tinel H, Cancela JM, Mogami H, Gerasimenko JV, Gerasimenko OV, Tepikin AV, and Petersen OH. Active mitochondria surrounding the pancreatic acinar granule region prevent spreading of inositol trisphosphate-evoked local cytosolic Ca2+ signals. EMBO J 18: 4999-5008, 1999. PMID: 10487752.
- Tomkotter L, Erbes J, Trepte C, Hinsch A, Dupree A, Bockhorn M, Mann O, Izbicki JR, and Bachmann K. The Effects of Pancreatic Microcirculatory Disturbances on Histopathologic Tissue Damage and the Outcome in Severe Acute Pancreatitis. Pancreas 45: 248-253, 2016. PMID: 26646271.
- Toth E, Maleth J, Zavogyan N, Fanczal J, Grassalkovich A, Erdos R, Pallagi P, Horvath G, Tretter L, Balint ER, Rakonczay Z, Jr., Venglovecz V, and Hegyi P. Novel mitochondrial transition pore inhibitor N-methyl-4-isoleucine cyclosporin is a new therapeutic option in acute pancreatitis. J Physiol 597: 5879-5898, 2019. PMID: 31631343.
- Valasani KR, Vangavaragu JR, Day VW, and Yan SS. Structure based design, synthesis, pharmacophore modeling, virtual screening, and molecular docking studies for identification of novel cyclophilin D inhibitors. J Chem Inf Model 54: 902-912, 2014. PMID: 24555519.
- van Dijk DPJ, Horstman AMH, Smeets JSJ, den Dulk M, Grabsch HI, Dejong CHC, Rensen SS, Olde Damink SWM, and van Loon LJC. Tumour-specific and organ-specific protein synthesis rates in patients with pancreatic cancer. J Cachexia Sarcopenia Muscle 10: 549-556, 2019. PMID: 30868736.
- Verma DD, Hartner WC, Levchenko TS, Bernstein EA, and Torchilin VP. ATP-loaded liposomes effectively protect the myocardium in rabbits with an acute experimental myocardial infarction. Pharm Res 22: 2115-2120, 2005. PMID: 16258743.
- Vinogradov AD. Steady-state and pre-steady-state kinetics of the mitochondrial F1Fo ATPase: is ATP synthase a reversible molecular machine? J Exp Biol 203: 41-49, 2000. PMID: 10600672.
- Voronina S, Collier D, Chvanov M, Middlehurst B, Beckett AJ, Prior IA, Criddle DN, Begg M, Mikoshiba K, Sutton R, and Tepikin AV. The role of Ca2+ influx in endocytic vacuole formation in pancreatic acinar cells. Biochem J 465: 405-412, 2015. PMID: 25370603.
- Voronina S, Longbottom R, Sutton R, Petersen OH, and Tepikin A. Bile acids induce calcium signals in mouse pancreatic acinar cells: implications for bile-induced pancreatic pathology. J Physiol 540: 49-55, 2002. PMID: 11927668.
- Voronina S, Sukhomlin T, Johnson PR, Erdemli G, Petersen OH, and Tepikin A. Correlation of NADH and Ca2+ signals in mouse pancreatic acinar cells. J Physiol 539: 41-52, 2002. PMID: 11850500.
- Voronina SG, Barrow SL, Simpson AW, Gerasimenko OV, da Silva Xavier G, Rutter GA, Petersen OH, and Tepikin AV. Dynamic changes in cytosolic and mitochondrial ATP levels in pancreatic acinar cells. Gastroenterology 138: 1976-1987, 2010. PMID: 20102715.
- Waldron RT, Chen Y, Pham H, Go A, Su HY, Hu C, Wen L, Husain SZ, Sugar CA, Roos J, Ramos S, Lugea A, Dunn M, Stauderman K, and Pandol SJ. The Orai Ca2+ channel inhibitor CM4620 targets both parenchymal and immune cells to reduce inflammation in experimental acute pancreatitis. J Physiol 597: 3085-3105, 2019. PMID: 31050811.
- Ward JB, Petersen OH, Jenkins SA, and Sutton R. Is an elevated concentration of acinar cytosolic free ionised calcium the trigger for acute pancreatitis? Lancet 346: 1016-1019, 1995. PMID: 7475553.
- Weidenbach H, Lerch MM, Gress TM, Pfaff D, Turi S, and Adler G. Vasoactive mediators and the progression from oedematous to necrotising experimental acute pancreatitis. Gut 37: 434-440, 1995. PMID: 7590444.
- Wen L, Voronina S, Javed MA, Awais M, Szatmary P, Latawiec D, Chvanov M, Collier D, Huang W, Barrett J, Begg M, Stauderman K, Roos J, Grigoryev S, Ramos S, Rogers E, Whitten J, Velicelebi G, Dunn M, Tepikin AV, Criddle DN, and Sutton R. Inhibitors of ORAI1 Prevent Cytosolic Calcium-Associated Injury of Human Pancreatic Acinar Cells and Acute Pancreatitis in 3 Mouse Models. Gastroenterology 149: 481-492 e7, 2015. PMID: 25917787.
- Williams JA. Cholecystokinin (CCK) Regulation of Pancreatic Acinar Cells: Physiological Actions and Signal Transduction Mechanisms. Compr Physiol 9: 535-564, 2019. PMID: 30873601.
- Williams JA, Korc M, and Dormer RL. Action of secretagogues on a new preparation of functionally intact, isolated pancreatic acini. Am J Physiol 235: 517-524, 1978. PMID: 215042.
- Yadava N, and Nicholls DG. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. J Neurosci 27: 7310-7317, 2007. PMID: 17611283.
- Yule DI. Ca2+ signaling in pancreatic acinar cells. Pancreapedia: Exocrine Pancreas Knowledge Base 2020. DOI: 10.3998/panc.2020.12.