
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2014.1
| Attachment | Size |
|---|---|
| 604.07 KB |
Introduction
Chronic pancreatitis is a progressive, inflammatory disease of the pancreas, which leads to inflammation and fibrosis, with the potential for exocrine and endocrine insufficiency. The prevalence of the disease is estimated to be 42 per 100,000 and rising (40,105). The pathogenesis of chronic pancreatitis is incompletely understood and complex, involving the interplay of environmental, metabolic, and genetic factors that modulate inflammatory and fibrotic pathways. Although alcohol is the leading cause of chronic pancreatitis, other toxic, hereditary, and obstructive insults can lead to disease with similar histologic and clinical manifestations. Studying the cellular mechanisms responsible for chronic pancreatitis is difficult in human subjects for a number of reasons, including the undefined natural history of the disease, lack of a reliable test for early chronic pancreatitis, and inaccessibility of human tissue. Therefore, animal models are essential to advancing our understanding of the pathophysiologic processes responsible for chronic pancreatitis.
Though chronic alcohol abuse is the leading cause of chronic pancreatitis, a number of other etiologies including toxins, obstructive lesions, and genetic disorders can cause chronic pancreatitis. There is no consensus as to how these diverse etiologic factors lead to a common histological endpoint. Theories of pathogenesis fall into two broad categories: those in which repeated episodes of acute pancreatitis lead to chronic pancreatitis and those in which an initiating injurious event is perpetuated and progresses to irreversible injury in the appropriate environment (Figure 1).

Figure 1. Models of evolution to chronic pancreatitis. In the traditional model of acute pancreatitis, repeated bouts of acute pancreatitis lead to fibrosis. An example of this model is the induction of chronic pancreatitis through repetitive cerulein injections. The Sentinal Acute Pancreatitis Event (SAPE) model proposes that an initiating event, like an episode of acute pancreatitis, activates the immune system allowing risk factors to drive a pro-fibrotic, anti-inflammatory pathways that lead to chronic pancreatitis. Ethanol sensitizations models, such as the ethanol/LPS model conform to this hypothesis. Both models of pancreatitis can result in similar severity of final pancreatic injury.
A prominent model of the latter is the sentinel acute pancreatitis event (SAPE) hypothesis, which posits that a single episode of acute pancreatitis can progress to chronic pancreatitis in a conducive environment. If the sentinel episode is severe enough to attract monocytes and to activate stellate cells, fibrosis can develop in the presence of continued injurious stimuli (99). Although some theories of injury focus on acinar cell injury, others focus on duct cells as initial target of damage (78, 104). Finally, some hypothesize that that multiple different pathways can lead to chronic pancreatitis using different mechanisms. All models of chronic pancreatitis, except autoimmune models, share the same histologic endpoints <b>(Figure 2)</b> and can lead to similar severity of disease. These include some or all of the following features: chronic inflammation, stellate cell proliferation/activation, acinar cell dropout, ductal dilatation, intraductal calcifications, and nerve enlargement.
Table 1. Chronic pancreatitis characteristics studied in animal models.

Features of chronic pancreatitis in human patients include multiple structural and functional characteristics that can be reproduced in animal models. No available animal model of chronic pancreatitis is known to replicate all of these features.

Figure 2. Histopathologic features of chronic pancreatitis. Animal models of chronic pancreatitis share histological endpoints, including fibrosis, pancreatic duct abnormalities, and cellular changes.

Figure 3. Animal models of chronic pancreatitis vary in mechanism and species used. A variety of different trigger mechanisms and animal species have been used in animal models of chronic pancreatitis.
Chemical Models
Cerulein pancreatitis
Various agents can be administered systemically or locally to produce chronic pancreatitis (Table 2).
Table 2. Chemical models of chronic pancreatitis.

Repetitive administration of cerulein, sometimes in the presence of sensitizing agents, is the most commonly used model of chronic pancreatitis. Ethanol can be combined with cerulein or LPS to produce chronic pancreatitis. Other toxic compounds can be administered systemically or by retrograde infusion into the pancreatic duct.
Treatment with supraphysiologic doses of the cholecystokinin (CCK) analogue, cerulein, is employed in a widely used animal model of acute pancreatitis. At low doses, cerulein provides physiologic stimulation to CCK receptors and enhances secretion from the acinar cell. However, animals treated with high dose cerulein (10-100 x physiologic doses; 20-50 μg/kg, intravenously or intraperitoneally) develop mild to moderate acute interstitial pancreatitis. The cerulein model of acute pancreatitis is characterized by aberrant zymogen activation in the acinar cell, inhibition of secretion, increased inflammation, and cellular damage. However, in this model of pancreatitis, there is recovery of exocrine pancreatic structure and function within 24 to 48 hours.
The cerulein model of chronic pancreatitis requires repeated cerulein injections over time and is the most commonly used, reproducible model of chronic pancreatitis. There are a number of protocols that vary in dose, interval, and duration of cerulein injections (23, 59, 106). This model produces morphohistologic findings compatible with chronic pancreatitis in humans, including fibrosis, chronic inflammation, atrophy, transdifferentiation of acini into duct-like cells, and ductal dilatation, (59) as seen in Figure 4.

Figure 4. Repeated cerulein injections lead to chronic pancreatitis in mice. After a two-week cerulein injection protocol, H&E (A) demonstrates significant inflammation and trichrome staining (B) reveals fibrosis in the exocrine pancreas. Images provided by Chuhan Chung, Yale University.
Interestingly, these findings can occur even in the absence of zymogen activation (77). This model is widely used because of its reproducibility and technical ease in rodents, making it an attractive technique for use in transgenic mice. The cerulein model mirrors chronic pancreatitis in humans because it involves repetitive injurious stimulation, paralleling the progression from recurrent acute pancreatitis to chronic pancreatitis that occurs in some patients (43). However, human pancreatitis is not associated with elevated levels of CCK. Furthermore, it remains controversial whether human acinar cells express CCK receptors, like rodent acinar cells (37, 58). Therefore, the relevance of the cerulein model to human disease, with respect to CCK’s role in human disease, can be questioned, though it may fully reflect other aspects of disease.
The cerulein model of chronic pancreatitis serves as the basis for studying the sensitizing effects of other agents. Lipopolysaccharide (LPS), a bacterial endotoxin, is a particularly relevant agent because chronic alcohol consumption leads to increased gut permeability, predisposing to bacterial translocation and increased serum LPS levels (7). LPS has been shown activate pancreatic stellate cells and stimulate inflammatory cytokines through activation of toll-like receptor 4 (TLR4) and nuclear factor κB (NFκB) (50). The addition of LPS to the repeated cerulein injection model accelerates the progression of disease and worsens its severity, measured by acinar cell atrophy, fibrosis, and the development of tubular complexes (62).
Cyclosporine A (CsA) has also been used a sensitizing agent in cerulein-induced chronic pancreatitis. In this model, rats received only two doses of intraperitoneal cerulein during a 15-day treatment with intraperitoneal CsA. Rats treated with cerulein alone recover fully from the acute cerulein pancreatitis, while those co-treated with cyclosporine exhibit chronic pancreatitis with atrophy, mononuclear inflammatory infiltrate, and enhanced collagen deposition (94). Increases in TGF-β are thought to mediate the effects of cyclosporine by activating pancreatic stellate cells, increasing the production of collagenase inhibitors, and inhibiting matrix degrading proteases. Due to reportedly high rates of toxicity by some investigators, the use of this model has been limited.
Other toxins
Feeding of a choline deficient ethionine-supplemented (CDE) diet induces acute hemorrhagic pancreatitis in mice (26, 60). The mechanism responsible for CDE-induced pancreatic damage is not known. Long-term administration of the CDE diet intermittently over 24 weeks leads to histological changes consistent with chronic pancreatitis including acinar atrophy, fibrosis, and the development of tubular complexes. Additionally, increased expression of EGFR, SPINK3, and TGF-α, which are all implicated in the pathogenesis from chronic pancreatitis to pancreatic adenocarcinoma were observed in this model. However, even after 54 weeks of CDE feeding, malignant lesions did not form (34).
L-arginine, an essential amino acids, administered intraperitoneally in high doses has been shown to cause severe, necrotizing acute pancreatitis in animal models (55). The mechanisms responsible for the effects of l-arginine on the pancreas are unknown, though production of reactive oxygen species and direct activation of the immune system have been postulated. Repeated injections of lower doses of l-arginine than cause severe acute disease over several weeks produce necrosis followed by chronic inflammation and fibrosis with impaired glucose tolerance in rats (98, 19). Unlike most human chronic pancreatitis, fibrotic tissue is replaced by adipose tissue over time, limiting its usefulness as a model of chronic pancreatitis histology.
Intravenous or intraperitoneal injection of dibutyltin dichloride (DBTC), a compound used in the production of polyvinyl chlorides, leads to acute interstitial pancreatitis through direct toxicity on the acinar cell and by causing chronic biliary obstruction through the formation of obstructing plugs in the distal common bile duct (51, 52). When repeated DBTC injections are administered, rats develop chronic inflammation and fibrosis (53, 85). However, this model is not highly reproducible, as only one third of animals display histological changes consistent with chronic pancreatitis. The CDE, l-arginine, and DBTC models are limited by the fact that none of these compounds cause pancreatitis in humans, their mechanisms of actions are unknown, and they each produce non-specific, extra-pancreatic injury.
Alcohol
Alcohol is the leading cause of chronic pancreatitis. However, less than 10% of alcoholics develop chronic pancreatitis (4). The lack of a homogeneous, dose-dependent effect of alcohol on the exocrine pancreas in humans is reflected in ethanol animal models of pancreatitis. Ethanol feeding alone does not produce chronic pancreatitis in animal models. The most informative use of this model has been its use in studies of sensitization to other injurious agents.
The Lieber-DeCarli method of chronic alcohol feeding involves supplementing liquid feeds with 36% ethanol and is useful in overcoming rodents’ natural aversion to alcohol (45). Pancreata from chronically alcohol fed animals appear histologically normal without significant gross inflammation or fibrosis. More intense alcohol feeding by continuous gavage, which produces sustained blood alcohol levels of 250-500 mg/dL, also fails to produce significant histological changes consistent with chronic pancreatitis (90). Other models of repeated ethanol feeding, including the ethanol agar block feeding model and supplementing drinking water with ethanol, fail to achieve consistently elevated levels of blood ethanol (6, 15). Therefore, chronic ethanol feeding cannot be used, on its own, as an experimental model of chronic pancreatitis. However, despite the lack of change in histology, chronic ethanol feeding has pronounced biochemical effects, including increased production of fatty acid esters, mitochondrial injury, and pancreatic stellate cell activation. Therefore, the model can still provide insight into the pathogenesis of alcohol induced chronic pancreatitis (44, 71, 102).
Combining chronic alcohol feeding with other injurious stimuli creates histologic damage and provides a reliable model of chronic pancreatitis. When rats fed the Lieber-DeCarli diet were challenged with repeated high-dose cerulein injections, findings consistent with human alcoholic chronic pancreatitis developed. While animals treated only with cerulein recovered, those sensitized with ethanol developed fibrosis, calcifications, and necrosis. Furthermore, the immune cell profile at each stage of injury was markedly different in ethanol-sensitized rats, compared to cerulein-only controls (21, 68). Similar results were seen in a mouse version of the same model (68).
In another model, rats fed Lieber-DiCarli diets were challenged with LPS. In this model ethanol-fed rats, but not rats on the control diet, developed fibrosis and displayed stellate cell activation (95). This model has direct clinical relevance because alcoholics are known to have increased gut permeability, which encourages higher circulating levels of LPS (7, 87). The LPS/alcohol model has been used to study the effects of alcohol abstinence on the progression of chronic pancreatitis and has shown that withdrawal of alcohol causes regression of fibrosis and increased pancreatic stellate cell apoptosis (94). Of the chronic pancreatitis summarized in this review, the combined use of ethanol feeding and LPS appears to have the most relevance to disease etiology encountered in clinical disease.
The histologic responses are also very similar to that which develops in human disease (Figure 5). These alcohol sensitization models conform to the SAPE hypothesis of chronic pancreatitis (Figure 1). Even in the presence of important risk factors like heavy alcohol intake, a sentinel inflammatory event, like an episode of acute pancreatitis, is required to activate the immune system and primes the pancreas for chronic inflammation. Then, depending on the presence of continued risk factors that modulate the immune response, fibrosis may develop (99).
Retrograde Infusion of Toxic Substances
Several models involving the retrograde infusion of toxic substances have been attempted. These models deliver toxins only to the pancreas, unlike the models that require systemic toxin administration described above. Retrograde infusion of the cytotoxic unsaturated fatty acid, oleic acid, destroys acinar cells and causes acute inflammation (57). Because the exocrine pancreatic parenchyma undergoes fatty replacement, rather than fibrosis over time, this model is not ideal for the study of human chronic pancreatitis. Infusion of trinitrobenzene sulfonic acid into the pancreatic duct leads to acute necrotizing pancreatitis at 48 hours and fibrosis, inflammation, and atrophy consistent with chronic pancreatitis at later time points (72). Retrograde infusion of bile acids provides an attractive model to study acute pancreatitis because gallstone obstruction is a common cause of acute pancreatitis (69). This method is thought to elicit pancreatitis through direct toxic effects on the acinar cell that is mediated by the bile acid receptor Gpbar1. Chronic pancreatitis caused by obstructive lesions may also be mediated by bile reflux into the pancreatic duct. While retrograde infusion of high doses of sodium taurocholate leads to death of most rats at 72 hours, some of the few survivors display atrophy and fibrosis (3). Retrograde infusion of lower doses of sodium taurocholate produces milder acute injury, with return to normal histopathology at 14 days (106). Drawbacks of this model include its technical difficulty and the finding that injury is often localized to the pancreatic head.

Figure 5. Alcohol/LPS leads to chronic pancreatitis in rats. In untreated rats (A) and rats treated with alcohol alone, acinar architecture is well-preserved and morphohistologic features of chronic pancreatitis are absent. In rats treated with alcohol in the presence of LPS (C,D), vacuolization (arrow), inflammatory infiltrate (arrowhead), acinar cell dropout, and fibrosis (blue staining in D) are observed, consistent with chronic pancreatitis. Images provided by Minoti Apte, University of New South Wales.
Obstructive Models
Obstructive lesions such as tumor, and trauma are rare causes of chronic pancreatitis. In humans with chronic pancreatitis from any cause, protein plugs form in small pancreatic ducts and pancreatic ductal pressure is elevated (9, 79). Therefore, obstruction of the pancreatic duct, provides a rational model for the study chronic pancreatitis (Table 3). One week after dogs and rats undergo ductal ligation, pancreata display ductal dilatation, disorganized acinar cell arrangement, fibrosis with collagen deposition, and inflammatory infiltrate in interstitial spaces (14).
Table 3. Mechanical models of chronic pancreatitis.

Obstructive chronic pancreatitis has been produced in several different species by complete or partial pancreatic duct ligation or through the induction of pancreatic ductal hypertension.
Pancreatic duct ligation in rabbits and dogs also results in significantly impaired glucose tolerance (33). Pancreatic ductal ligation in the mouse is technically challenging because of the small size of the pancreatic duct. Furthermore, the redundant duct anatomy of the mouse leads to histopathological changes in some, but not all, pancreatic lobes. Edema, increased inflammatory infiltrate, enhanced apoptosis, and acinar cell dropout are seen three days after pancreatic duct ligation (81). By five days after pancreatic ductal ligation, there few remaining normal acinar cells and abundant proliferation of duct-like cells with fibrosis. Seven days after ligation, metaplastic ducts, which are resistant to apoptosis are observed. Figure 6 demonstrates the early histological changes after pancreatic duct ligation. Several months after pancreatic duct ligation, there is intralobular fatty replacement of the exocrine pancreas (97). Partial pancreatic obstruction in dogs can be accomplished by inserting a small plastic tube into the pancreatic duct. This model produces pancreatic acinar atrophy, fibrosis, and inflammation (88).

Figure 6. Pancreatic ductal ligation leads to histology consistent with chronic pancreatitis in mice. Characteristic histological features including loss of normal acinar architecture, inflammation, and formation duct-like structures (arrows) are seen soon after pancreatic duct ligation in the mouse. Images provided by Howard Crawford, Mayo Clinic (Jacksonville Florida).
Given that pancreatic duct ligation leads to eventual complete loss of acinar cells, rather than fibrosis, a model that causes pancreatic ductal hypertension without pancreatic ductal obstruction was developed. In this technically challenging model, pancreatic ductal hypertension was induced by implanting pancreatic, biliary, and duodenal cannulas in rats. The free end of the pancreatic duct cannula was then vertically raised to create increased hydrostatic pressure for two weeks (106). In rats with pancreatic ductal hypertension, there was significant fibrosis with collagen deposition, lymphocytic inflammatory infiltrate, and plug formation in the main pancreatic duct.
Genetic Models
Several transgenic models that are based on known susceptibility genes in humans have been developed (Table 4). Therefore, these animal models have direct clinical relevance to human disease. The fidelity with which the phenotype of the transgenic animal models human disease varies widely.
Table 4. Genetic models of chronic pancreatitis.

Genetic models of chronic pancreatitis can target genes that are generally expressed or specifically expressed in acinar or duct cells.
CFTR
Cystic fibrosis is a common autosomal recessive disease among Caucasian populations that affects multiple organ systems, including the respiratory tract and gastrointestinal organs. It is caused by mutations in the CFTR gene located on chromosome 7, which encodes a regulated chloride channel. Most patients with cystic fibrosis develop pancreatic insufficiency. Patients with pancreatic sufficiency are at high risk for chronic pancreatitis (18). Patients who are not homozygotes or compound heterozygotes for the classical, severe CFTR mutations and, therefore, do not have cystic fibrosis, can express other milder phenotypic variants. Patients who are homozygotes or compound heterozygotes, with at least one allele encoding a mild variant have a 40-80% increased risk of developing chronic pancreatitis (66). Heterozygotes for CFTR mutations have a 3-4 fold increased risk of developing chronic pancreatitis, with the subset of patients who co-express mutated SPINK1 (serine protease inhibitor Kazal type 1) at the highest risk (98).
Several transgenic animal models have been used to study the relationship between CFTR mutations and chronic pancreatitis. Mice homozygous for the S489X mutation, which encodes a truncated protein, display a phenotype that has many features of human cystic fibrosis (84). In particular, these mice develop failure to thrive, meconium ileus, and alterations in mucous and serous glands. The mice have modest exocrine insufficiency, with lower trypsin and lipase activity than controls, an abnormally acidic duodenum because of decreased pancreatic bicarbonate secretion, and blunted secretory responses to CCK (20). CFTR mutant mice develop more severe acute cerulein-induced pancreatitis with a more exuberant inflammatory response and decreased apoptosis than controls (22). Although these transgenic mice have enhanced expression of pro-inflammatory cytokine genes at baseline, they do not develop chronic pancreatitis. Death from intestinal obstruction occurs during weaning by 40 days of age in nearly all CFTR -/- mice, limiting the usefulness of this murine model for studying CFTR-related chronic pancreatitis.
In an effort to more closely replicate human disease, a porcine model of cystic fibrosis was developed. CFTR -/- pigs appear normal at birth, but soon develop meconium ileus and failure to thrive (75). The piglets require surgery to relieve intestinal obstruction from meconium ileus and prevent perforation. They also eventually develop infertility and focal biliary cirrhosis. Porcine CFTR -/- pancreata appeared small, with increased adiposity and inflammation. Centroacinar spaces and ducts were dilated and obstructed by eosinophilic material. CF pigs also have significantly lower levels of pancreatic amylase, lipase, and trypsin (91). The baseline volume and pH of pancreatic fluid is depressed in CF pigs, and they do not respond to secretin stimulation with increased pancreatic secretions like wild type pigs. Newborn CF piglet pancreata display a mixed inflammatory infiltrate with neutrophils, macrophages, effector and cytotoxic T cells, activated, T helper cells, and natural killer T cells (2). Additionally, fetal CF pig acinar cells display increased expression of proinflammatory, complement cascade, and profibrotic genes. Furthermore, increased apoptosis, α-smooth muscle actin, and TGFβ-1 were seen in newborn and fetal CF pigs. These findings suggest of upregulation of fibrotic pathways, providing a superior model for CF-related pancreatic disease (1). Although the CF pig model appears to very closely resemble human disease, the high cost of the animals limits the opportunities for studying this model.
A ferret model of CF has also been developed. Newborn CFTR -/- animals displayed dilated pancreatic ducts with inspissated zymogen secretions. Seventy-five percent of animals maintained this phenotype during infancy. A small minority of cases developed more severe lesions, including fibrosis and loss of pancreatic parenchyma. Because these animals have high early mortality because of meconium ileus and GI malabsorption, a gut-corrected transgenic CFTR-knockout, which expresses CFTR only in the intestines and does not suffer from meconium ileus, was created (86). Although the latter model is more amenable to study, its use has been limited.
PRSS1
Mutations in cationic trypsinogen are responsible for 80% of cases of hereditary pancreatitis that are not caused by cystic fibrosis. These mutations affect the regulatory regions of trypsinogen, rendering it more vulnerable to inappropriate activation. The R122H missense mutation of the cationic trypsinogen gene (PRSS1) was the first identified mutation responsible for hereditary pancreatitis (100) and is an attractive target for manipulation in mouse models. In one model, transgenic mice with the R122H mutation in murine trypsin 4 targeted to the pancreatic acinar cell were produced. These mice developed inflammation, fibrosis, and acinar cell dedifferentiation. There was activation of acinar cell-specific inflammatory signaling pathways and c-jun-N-terminal kinase, which mediates TNF-α induced cell death (5). Unfortunately, this model is no longer available. Another model created mice transgenic for human R122H cationic trypsinogen, targeted to the pancreas. These animals had elevated lipase levels, but no spontaneous alterations in pancreatic histology (82). In both models, cerulein-induced chronic pancreatitis was more severe.
SPINK3
The serine protease inhibitor Kazal type 1 gene (SPINK1) encodes for a protease inhibitor that is upregulated in inflammatory states and safeguards against aberrant intracellular zymogen activation. SPINK mutations are common in humans, though the vast majority of those with even “high risk” mutations do not develop pancreatitis (70). Deficiency of SPINK3, a homologue of SPINK1, is lethal by two weeks in knockout mice. When acinar cells isolated from neonatal mice were analyzed, enhanced trypsin activation was identified (64). Embryonic pancreatic specimens revealed autophagic degeneration of acinar cells, which progressed to rapid cell death after birth (65). SPINK3 heterozygotes have reduced trypsin inhibitor capacity than wild type mice because they express less SPINK3 protein. However, SPINK3 heterozygotes do demonstrate increased susceptibility to cerulein-induced pancreatitis, suggesting that a threshold level of SPINK3 is sufficient to protect against pancreatitis (76). Although these animals do not manifest spontaneous chronic pancreatitis, the model highlights the important role of SPINK in the maintenance and regeneration of the exocrine pancreas.
Kras
The oncogene Kras is the most common gene mutated in pancreatic ductal adenocarcinoma (PDAC) and is also mutated in about 40% of patients with chronic pancreatitis (47). Transgenic mice with overexpression of activated Kras in acinar cells demonstrated fibrosis and inflammation that mimicked the histologic findings of human chronic pancreatitis (38). Elevated levels of Ras activity, in this model, led to the spontaneous development of PDAC, making it an attractive model for the progression from chronic pancreatitis to pancreatic cancer. Inflammatory stimuli that produce transient Ras signaling in wild type animals induced prolonged Ras, NF-κB, and COX-2 activity in mice expressing oncogenic Kras, leading to chronic inflammation and precancerous lesions (17). Thus, the Kras model likely reflects a mechanism seen in human chronic pancreatitis with respect to chronic inflammation and fibrosis.
Other Genetic Models
Several other genetic models do not have an obvious correlate in human disease, but can be useful in understanding the pathways that lead to chronic pancreatitis. Acinar cells have an extensive endoplasmic reticulum network with associated chaperones and foldases to manage the production and secretion of digestive enzymes. Regulatory mechanisms direct misfolded proteins to ER-associated degradation pathways. Stressors such as oxidative damage, overloading the protein folding capacity of the ER, or the presence of mutant proteins lead to ER stress and trigger the unfolded protein response. The unfolded protein response is an important mediator of acinar cell damage in alcohol-induced pancreatitis (48). The ER sensor, PERK (protein kinase RNA-like ER kinase), responds to ER stress by decreasing overall protein translation while enhancing regulators of redox status and glutathione production. Mice with pancreas-specific PERK knockout develop pancreatic exocrine and endocrine dysfunction rapidly after birth (31). Acinar cell death in this model occurs through ischemia, which then triggers an inflammatory response consisting of neutrophils and macrophages (36). This model highlights the importance of the ER stress response in maintaining acinar cell function and may be relevant to the rare human disease Wolcott-Rallison syndrome, which is caused by a mutation in the PERK gene and characterized by early onset diabetes, skeletal dysplasia, and exocrine pancreatic insufficiency .
Defects in primary cilia have been implicated in polycystic kidney disease (PKD). Pancreatic lesions, especially pancreatic cysts and occasionally chronic pancreatitis, are more common in patients in PKD. To study this disease, a mouse model with pancreas-specific inactivation of Kif3a, which encodes kinesin-2, was developed. Kinesin-2 is a protein that is required for cilia assembly. The pancreata of these mice display acinoductular metaplasia, fibrosis, eventual pancreatic lipomatosis, and cyst formation (10). These findings provide a model for the pancreatic diseases associated with PKD and primary ciliary dyskinesia (Kartagener’s syndrome).
Another model focuses on the role of the inflammatory cytokine IL-1β. In this model, transgenic mice that selectively overexpress human IL-1β in the pancreas were studied. These elastase sshIL-1β mice develop acinar cell atrophy, pancreatic ductal dilatation, mixed inflammatory infiltrate, acinar-ductal metaplasia, and fibrosis (49). The mice were also susceptible to more severe chronic pancreatitis after 20 weeks of cerulein treatment. There was increased expression of tumor necrosis factor-alpha (TNF-α), chemokine (C-X-C motif) ligand 1, stromal cell-derived factor 1, transforming growth factor-beta1 (TGF-β1), matrix metallopeptidase 2, 7, and 9, inhibitor of metalloproteinase 1, and cyclooxygenase 2 in the model. Given this profile, IL-1β likely leads acinar cell damage by recruiting and activating inflammatory cells and pancreatic stellate cells.
WBN/Kob rats, initially developed as a model for gastric tumors, also spontaneously develop pancreatic fibrosis and diabetes mellitus (61). By three months of age, the mice develop focal pancreatic necrosis and inflammation that slowly extends to encompass the entire pancreas. By four months, fibrosis is seen in the exocrine pancreas, with eventual expansion to the endocrine pancreas. The rats become diabetic at 60-90 months. These finding are only seen in sexually mature, male mice, suggesting a role for androgen in the pathogenesis of the pancreatic lesions. A limitation of this model is that exact nature of the genetic alterations responsible for the phenotype of this mouse strain is unknown. Chromosomal mapping identified polymorphisms in three candidate genes, Rac2, Grap2, and Xpnpep3 (56). Rac2 is a GTPase member of the Rho family, which plays a role in apoptosis, phagocytosis, and cytoskeletal reorganization. Grap2 is an adaptor protein that participates in leukocyte specific protein-tyrosine kinase signaling. Finally, Xpnpep3, localizes to mitochondria and is involved in ciliary function. Which of these candidate genes is responsible for the pancreatic phenotype of WBN/Kob mice requires further study.
The hedgehog signaling pathway plays a key role in patterning events in normal mammalian development. Indian hedgehog (Ihh), a member of this family, is upregulated in human chronic pancreatitis (41). To explore the contribution of the Hedgehog pathway in the pathogenesis of chronic pancreatitis, transgenic zebrafish that overexpress Ihh and Sonic hedgehog (Shh) along with green fluorescent protein were developed (39). Both Ihh and Shh mutants had similar phenotypes. Neither transgenic zebrafish developed derangements in acinar differentiation, but both developed fibrosis over time with increased expression of matrix metalloproteinase and TGF-β.
Keratins are epithelia-specific intermediate filament proteins involved in pancreatic acinar cell homeostasis. In humans, some studies suggest that mutations in the keratin 8 gene are associated with chronic pancreatitis, (12) but others did not find an association between keratin 8 mutations and chronic pancreatitis (89). When the human keratin K8 is overexpressed in HK8 transgenic mice, acinar cells display inflammation, fibrosis, dysplasia, and parenchymal replacement by adipose tissue (11).
The E2F family of DNA-binding transcriptional activators is comprised of six members (E2F1-6) that heterodimerize with the transcription factor DP to regulate the expression of genes involved in cell growth and differentiation. E2F1/E2F2 double deficient (DKO) mice lose acinar cells, develop fibrosis and fat replacement, without significant inflammation after two weeks. The endocrine pancreas also became progressively atrophic. The mice have a shortened lifespan, in part, because of the development of frank diabetes with decreased insulin levels and hyperglycemia and exocrine insufficiency with steatorrhea. These studies suggest that E2F1 and E2F2 play a critical role in the maintenance of pancreatic homeostasis (35).
IκB kinase α (IΚΚα), a subunit of the IΚΚ complex, regulates the activation of the NF-κΒ transcription factor and is critical for epidermal differentiation, keratinocyte differentiation, and skeletal patterning. Conditional IΚΚα knockout mice, with IΚΚα deficiency in acinar, ductal, and islet cells develop spontaneous and progressive acinar cell damage, fibrosis, and inflammation. Additionally, mice develop endocrine insufficiency. The pathway underlying these defects was shown to be impaired autophagic protein degradation, leading to ER stress and elevated expression of CHOP (C/EBP homologous protein), a proapoptotic transcription factor (46).
Pigment epithelial-derived factor (PEDF) is involved in maintaining a normal extracellular matrix and regulation of intracellular lipid metabolism. At baseline, PEDF-null mice express markers suggestive of pancreatic stellate cell activation, but no changes in acinar cell morphology. However, there is sensitization to cerulein-induced chronic injury. PEDF-knockout animals displayed more pronounced fibrotic changes than wild-type mice with greater weight loss, suggestive of exocrine insufficiency. Surprisingly, the PEDF-deficient mice recovered from fibrotic injury similarly to wild type controls, suggesting the PEDF is not required for the compensatory mechanisms involved in the resolution of pancreatic tissue fibrosis (80).
Pancreatic duct cell
Cyclooxygenase-2 (COX-2) is a rate-limiting enzyme for the production of prostaglandin, which is a critical mediator of chronic inflammation. Mice with overexpression of COX-2 driven by a BK5 promoter (BK5 mice) have high levels of COX-2 in ductal cells. High prostaglandin levels drive the development of chronic pancreatitis, with histologic features including inflammation, fibrosis, and ductal metaplasia foci. Older mice spontaneously develop lesions consistent with pancreatic ductal adenocarcinoma (16).
Liver X receptor β (LXRβ) is a nuclear receptor with a key role in cholesterol, triglyceride, and glucose metabolism and is expressed in pancreatic duct epithelial cells. LXRβ null mice display periductal inflammation with increased cell death of ductal epithelial cells. Additionally, pancreatic cells have enlarged golgi cisternae and the ducts are dilated with markedly dense secretory fluid. The knockout mice develop pancreatic exocrine insufficiency with weight loss and decreased fat stores. The secretory defect in LXRβ null mice is likely mediated by loss of aquaporin-1, a membrane water channel protein that regulates transcellular fluid transport (24).
Viral Models
Infection with group B coxsackieviruses has been implicated in a number of diseases including acute and chronic pancreatitis, making it a clinically relevant model of disease (74). This model of pancreatitis involves infection with one of two coxsackie B4 virus strains. The CVB4-P strain produces mild acute pancreatitis, which completely resolves after ten days. The more virulent CBV4-V strain produces more severe, necrotizing acute pancreatitis, followed by a chronic phase of disease characterized by acinar-ductal metaplasia, an inflammatory infiltrate, fibrosis, and fatty replacement of the pancreas. Microarray data analysis was employed to determine which genes correlated with disease resolution versus progression to chronic disease. In mild, reversible CVB4-P disease, there is enhanced expression of embryonic markers, which are likely involved in pancreatic regeneration. The gene expression map for the chronic CVB4-V model emphasized genes involved in apoptosis and fibrosis. Markers of innate and adaptive immunity also varied in the two models. While the CVB4-P infection is associated with alternatively activated (M2) macrophages and T helper 2 (Th2) cells, the progressive CVB4-V model is characterized by increased expression of classically activated (M1) macrophages and T helper 1 (Th1). The differences between CVB4-P and CVB4-V infection provide a useful approach to study why some injurious stimuli lead to reversible pancreatic injury in humans, while others progress to chronic pancreatitis (67).
Models of Autoimmune Pancreatitis
Autoimmune pancreatitis (AIP) is a distinct fibro-inflammatory pancreatic disorder with two subtypes. Type 1 AIP is associated with elevated serum IgG4 and IgG4 positive lesions in other tissues. Additionally, the pancreas of patients affected by Type 1 is often diffusely enlarged with characteristic lymphoplasmacytic sclerosing histology. In contrast, Type 2 AIP is not associated with IgG4. Instead, the exocrine pancreas develops focal granulocyte-epithelial lesions. Type 2 AIP is associated with inflammatory bowel disease in more that 15% of cases.
MRL-Mpmice spontaneously develop a number of autoimmune diseases including glomerulonephritis, arteritis, and arthritis. Chronic pancreatitis develops in 75% of female MRL-Mp mice at 34-38 weeks with mononuclear inflammatory infiltrate, destruction and fatty replacement of pancreatic acini (42). The injection of polyinosinic:polycytidylic acid (poly I:C), which is structurally similar to double stranded viral RNA and pro-inflammatory, leads to activation of macrophages, NK and B cells, and increased cytokine production, accelerating and improving the penetrance of disease. All female MRL-Mp mice treated with poly I:C develop chronic pancreatitis with infiltration of CD4+ T cells and activated macrophages by 18 weeks. In this model, the mice do not develop other overt auto-immune conditions (73).
Interleukin 10–deficient (IL-10KO) mice spontaneously develop colitis and are a widely used model of inflammatory bowel disease. When IL-10KO mice were treated with poly I:C, they develop pancreatitis with a mononuclear inflammatory infiltrate, destruction of acini, fibrosis, and fatty changes (108). This histology, like that of the MRL-Mp model, resembles that seen with type I AIP. This finding is unexpected because type 2 AIP, and not type 1, is associated with inflammatory bowel disease.
One theory of the pathogenesis of AIP implicates immune responses to microorganisms in the environmental etiology of AIP. To explore this hypothesis, mice were injected with heat-killed E. coli. Shortly after inoculation, these mice developed acinar inflammatory infiltrate composed mainly of granulocytes and periductal fibrosis. Months after the final inoculation, lesions similar to granulocyte epithelial lesions of type 2 AIP formed. There was also marked, periductal fibrosis and acinar to ductal metaplasia. E. Coli treated mice also had increased serum IgG levels and extra-pancreatic involvement of disease with salivary gland involvement. This model mixes features of type I AIP (extra-pancreatic involvement, elevated IgG) and type 2 AIP (histology) (32).
A lesser used model immunizes neonatally thymectomized mice with carbonic anhydrase and lactoferrin. These agents were chosen because autoantibodies against carbonic anhydrase II and lactoferrin have been identified in patients with AIP. These mice developed pancreatic inflammation that was mediated by Th1 CD4+ T cells (92). Additionally, there was extra-pancreatic involvement with salivary gland inflammation and cholangitis.
Though each of these models employs an autoimmune pathogenic mechanism, none of the models accurately reproduce the constellation of clinical and histological features of either type 1 or type 2 AIP. Therefore, the relevance to human disease is unclear. Another issue complicating this and all murine models of inflammatory disease is the lack of congruence of genomic responses to inflammatory stimuli in humans and mice (83).
Clinical Relevance of Models of Pancreatitis
Selecting the appropriate animal model of chronic pancreatitis depends on the experimental question being investigated. Table 5 summarizes the most relevant features of the various models of chronic pancreatitis. The most widely used animal model of chronic pancreatitis is the repetitive cerulein injection model. This non-surgical model allows for a relatively technically simple, inexpensive, flexible, and highly reproducible mechanism of replicating the histopathologic findings seen in human pancreatitis. However, given that hyperstimulation is not involved the pathogenesis of human disease, the relevance of the cerulein model with respect to disease initiation is questionable, though it does accurately represent later events in human disease. The LPS/ethanol model of pancreatitis is a clinically relevant model of the events that lead to alcoholic chronic pancreatitis. It is also unique in its ability to address the durability/reversibility of early disease. However, chronic alcohol feeding of rodents can be challenging. Additionally, it is a rat model and therefore not optimized for genetically modified mice. Another complicating factor in all mice models is that recent studies comparing acute inflammatory responses between mice and humans have shown dramatic differences in cytokine responses between these two species (83). The types of inflammatory cells and cytokines expressed in animal models of chronic pancreatitis, their levels, and time-dependent are being defined. Studies to date have been performed in only a few models (67, 75) and underscore the complexity of these responses. It will be a challenge to determine whether these responses, which are likely central to the pathogenesis of chronic pancreatitis, are conserved between species. Another issue with animal models of pancreatitis is that, although they provide an accurate morphohistologic model of pancreatitis, they do not fully address other clinically relevant aspects of human disease, including endocrine and exocrine insufficiency, pain, and increased susceptibility to pancreatic adenocarcinoma.
Exocrine and endocrine insufficiency occur late in the course of chronic pancreatitis. The extent of exocrine insufficiency produced by the cerulein model of chronic pancreatitis is not well characterized. Though, the significant decrease in acinar cell protein content at six weeks may suggest a reduction in exocrine functioning (63). The cerulein model does not produce endocrine insufficiency on its own. However, when combined another insult, the cerulein model can result in endocrine insufficiency. In one rat model, the repetitive cerulein injection model is combined with the stress of water immersion (27, 54). This model results in histologic endocrine cellular damage, hyperglycemia, and decreased insulin levels. The l-arginine model results in exocrine and endocrine insufficiency coinciding with pancreatic atrophy (97). The WBN/Kob mouse model also produces endocrine insufficiency, with diabetes occurring spontaneously at 60-90 weeks. PERK (-/-) mice have a decrease in endocrine function between four and eight weeks (31, 61). Both WBN/Kob and PERK (-/-) mice also exhibit exocrine insufficiency. Additionally, the surgical pancreatic duct ligation models produce exocrine and endocrine insufficiency (8, 88).
Chronic pancreatitis is an important risk factor for the development of pancreatic adenocarcinoma, increasing the risk of cancer tenfold. The potential for progression from pancreatitis to cancer is not addressed in the most commonly used animal models. Mouse models, in which oncogenic Kras is overexpressed provide a useful, clinically relevant model of the development of pancreatic adenocarcinoma from chronic pancreatitis. In experimental models utilizing oncogenic Kras, healthy cells are resistant to malignant transformation, while a background of chronic pancreatitis encourages high penetrance of oncogenic Kras with the development of PanINs and PDAC (29, 30). It is likely that chronic pancreatic injury induces acinar cell proliferation for tissue repair, either by dedifferentiation of mature acinar cells or recruitment of progenitor cells (28). These cells, which have increased embryonic markers, may be key in the progression from pancreatitis to cancer.
Abdominal pain in acute pancreatitis can be severe, impacts patients’ quality of life, and is the most frequent reason for hospitalization (25). Given that there are no targeted therapies for chronic pancreatitis pain, characterization of pain pathways could be helpful in developing potential therapies. In mice treated with the DBTC model of pancreatitis, pain as measured by behavioral responses to visceral stimuli, was mediated by bradykinin and IL-6 (13, 93). The trinitrobenzene sulfonic acid infusion model identified nerve growth factor as a mediator of pancreatic nociceptor excitability and pain behaviors in rats (101, 109, 110).
Conclusion
Animal models provide a reproducible method for examining the pathogenesis of chronic pancreatitis. Repetitive cerulein injection is the most widely used model because it is technically straightforward, reproducible, and flexible. However, while the pancreatic histopathologic findings reproduce those seen in humans with chronic pancreatitis, the relevance of cerulein as a triggering mechanism in the pathogenesis of human disease is questionable. Other models, like the alcohol/LPS model, mechanical injury, genetic, and viral models employ triggers with direct correlates to known risk factors for chronic pancreatitis in humans. Ultimately, the choice of animal model depends upon the hypothesis being tested. A central concern in unraveling the mechanisms responsible for chronic pancreatitis in humans is understanding how genetic and environmental risk factors interact to initiate and advance disease in some patients but not others. This issue presents a particular challenge in the development of animal models, which must be highly reproducible to be practically useful. Furthermore, no currently available models reproduce all relevant aspects of disease—histopathology, endocrine/exocrine insufficiency, pain, increased risk of progression to cancer. The development of new models or combining known models in new ways may provide further insight into the complex pathogenesis of chronic pancreatitis.
Table 5. Summary of animal models of chronic pancreatitis.
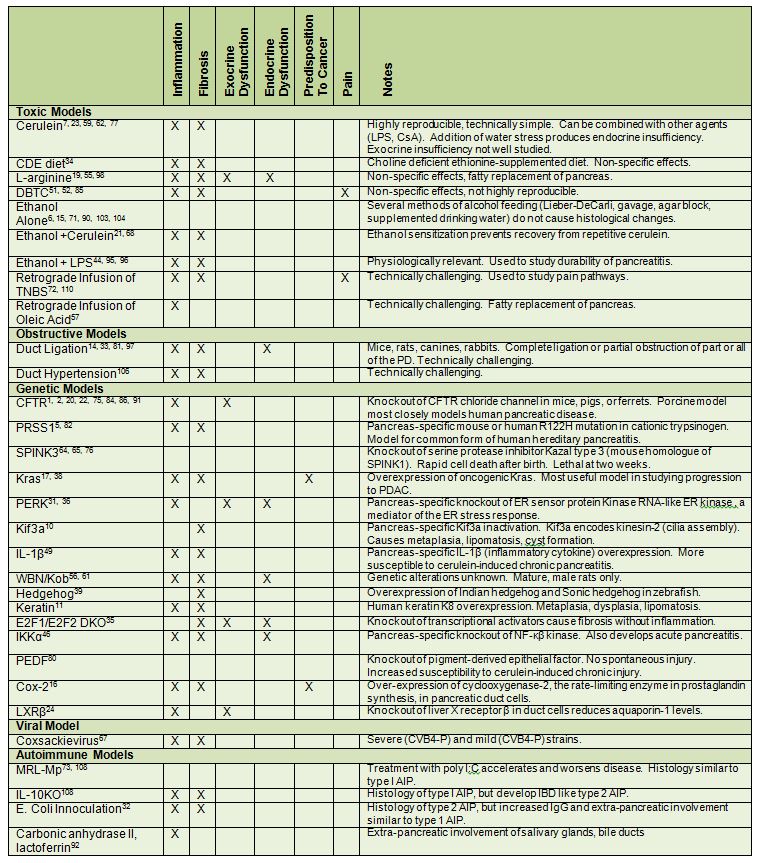
Models of pancreatitis are classified by their mechanism of injury and clinically relevant features.
Funding:
Supported by a National Institutes of Health K08 (DK090104 ) to A.M.R and a Merit Award from the Veterans Administration, and the National Institutes of Health RO1 (DK54021) from NIDDK, and the National Institutes of Health R21 (AA020847) from NIAAA to F.S.G.
References
- Abu-El-Haija M, Ramachandran S, Meyerholz D.K, Abu-El-Haija M, Griffin M, Giriyappa R.L, Stoltz D.A, Welsh M.J, McCray P.B, Jr, Uc A. Pancreatic damage in fetal and newborn cystic fibrosis pigs involves the activation of inflammatory and remodeling pathways. Am J Patho 181: 499-507, 2012. PMID: 22683312
- Abu-El-Haija M, Sinkora M, Meyerholz D.K, Welsh M.J, McCray P.B. Jr, Butler J, Uc A. An activated immune and inflammatory response targets the pancreas of newborn pigs with cystic fibrosis. Pancreatology 11: 506-515, 2011. PMID: 22057257
- Aho H.J, Koskensalo S.M, Nevalainen T.J. Experimental pancreatitis in the rat. Sodium taurocholate-induced acute haemorrhagic pancreatitis. Scand J Gastroenterol 15: 411-416, 1980. PMID: 7433903
- Ammann, R.W. The natural history of alcoholic chronic pancreatitis. Intern Med 40: 368-375, 2001. PMID: 11393404
- Archer H, Jura N, Keller J, Jacobson M, Bar-Sagi D. A mouse model of hereditary pancreatitis generated by transgenic expression of R122H trypsinogen. Gastroenterology 131: 1844-1855, 2006. PMID: 17087933
- Bautista A.P. Chronic alcohol intoxication induces hepatic injury through enhanced macrophage inflammatory protein-2 production and intercellular adhesion molecule-1 expression in the liver. Hepatology 25: 335-342, 1997. PMID: 9021944
- Bode C, V. Kugler, J.C. Bode. Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J Hepatol 4: 8-14, 1987. PMID: 3571935
- Boerma D, Straatsburg I.H, Offerhaus G.J, Gouma D.J, Van Gulik T.M. Experimental model of obstructive, chronic pancreatitis in pigs. Dig Surg 20: 520-526, 2003. PMID: 14534374
- Bradley E.L. 3rd, Pancreatic duct pressure in chronic pancreatitis. Am J Surg 144: 313-316, 1982. PMID: 7114368
- Cano D.A, Sekine S, Hebrok M. Primary cilia deletion in pancreatic epithelial cells results in cyst formation and pancreatitis. Gastroenterology 131: 1856-1869, 2006. PMID: 17123526
- Casanova M.L, Bravo A, Ramirez A, Morreale de Escobar G, were F, Merlino G, Vidal M, Jorcano J.L. Exocrine pancreatic disorders in transsgenic mice expressing human keratin 8. J Clin Invest 103: 1587-1595, 1999. PMID: 10359568
- Cavestro G.M, Frulloni L, Nouvenne a, Neri T.M, Calore B, Ferri B, Bovo P, Okolicsanyi L, Di Mario F, Cavallini G. Association of keratin 8 gene mutation with chronic pancreatitis. Dig Liver Dis 35: 416-420, 2003. PMID: 12868678
- Chen Q, Vera-Portocarrero L.P, Ossipov M.H, Vardanyan M, Lai J, Porreca F. Attenuation of persistent experimental pancreatitis pain by a bradykinin b2 receptor antagonist. Pancreas 39: 1220-1225, 2010. PMID: 20531238
- Churg A, W.R. Richter. Early changes in the exocrine pancreas of the dog and rat after ligation of the pancreatic duct. A light and electron microscopic study. Am J Pathol 63: 521-546, 1971. PMID: 5581235
- Coleman R.A, Young B.M, Turner L.E, Cook R.T. A practical method of chronic ethanol administration in mice. Methods Mol Biol 447: 49-59, 2008. PMID: 18369910
- Colby J.K, Klein R.D, McArthur M.J, Conti C.J, Kiquchi K, Kawamoto T, Riggs P.K, Pavone A.L, Sawicki J, Fischer S.M. Progressive metaplastic and dysplastic changes in mouse pancreas induced by cyclooxygenase-2 overexpression. Neoplasia 10: 782-796, 2008. PMID: 18670639
- Daniluk J, Liu Y, Deng D, Chu J, Huang H, Gaiser S, Cruz-Monserrate Z, Wang H, Ji B, Logsdon C.D. An NF-kappaB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J Clin Invest 122: 1519-1528, 2012. PMID: 22406536
- De Boeck K, Weren M, Proesmans M, Kerem E. Pancreatitis among patients with cystic fibrosis: correlation with pancreatic status and genotype. Pediatrics 115: e463-e469, 2005. PMID: 15772171
- Delaney C.P, McGeeney K.F, Dervan P, Fitzpatrick J.M. Pancreatic atrophy: a new model using serial intra-peritoneal injections of L-arginine. Scand J Gastroenterol 28: 1086-1090, 1993. PMID: 8303212
- De Lisle R.C, Isom K.S, Ziemer D, Cotton C.U. Changes in the exocrine pancreas secondary to altered small intestinal function in the CF mouse. Am J Physiol Gastrointest Liver Physiol 281: G899-G906, 2001. PMID: 11557509
- Deng X, Wang L, Elm M.S, Gabazadeh D, Diorio G.J, Eagon P.K, Whitcomb D.C. Chronic alcohol consumption accelerates fibrosis in response to cerulein-induced pancreatitis in rats. Am J Pathol 166: 93-106, 2005. PMID: 15632003
- Dimagno M.J, Lee S.H, Hao Y, Zhou S.Y, McKenna B.J, Owyang C. A proinflammatory, antiapoptotic phenotype underlies the susceptibility to acute pancreatitis in cystic fibrosis transmembrane regulator (-/-) mice. Gastroenterology 129: 665-681, 2005. PMID: 16083720
- Feng D, Park O, Radaeva S, Wang H, Yin s, Kong X, Zheng M, Zakhari S, Kolls J.K, Gao B.. Interleukin-22 ameliorates cerulein-induced pancreatitis in mice by inhibiting the autophagic pathway. Int J Biol Sci 8: 249-257, 2012. PMID: 22253568
- Gabbi C, Kim H.J, Hultenby K, Bouton D, Toresson G, Warner M, Gustagsson J.A. Pancreatic exocrine insufficiency in LXRbeta-/- mice is associated with a reduction in aquaporin-1 expression. Proc Natl Acad Sci U S A 105: 15052-15057, 2008. PMID: 18806227
- Gardner T.B, Kennedy A.T, Gelrud A, Banks P.A, Vege S.S, Gordon S.R, Lacy B.E. Chronic pancreatitis and its effect on employment and health care experience: results of a prospective American multicenter study. Pancreas 39: 498-501, 2010. PMID: 20118821
- Gilliland L, M.L. Steer. Effects of ethionine on digestive enzyme synthesis and discharge by mouse pancreas. Am J Physiol 239: G418-G426, 1980. PMID: 6159794
- Goto M, Nakano I, Kimura T, Miyahara T, Kinjo M, Nawata H. New chronic pancreatitis model with diabetes induced by cerulein plus stress in rats. Dig Dis Sci 40: 2356-2363, 1995. PMID: 7587814
- Guerra C, Barbacid M. Genetically engineered mouse models of pancreatic adenocarcinoma. Mol Oncol 7: 232-247, 2013. PMID: 23506980
- Guerra C, Collado M, Navas C, Schuhmacher A.J, Hernandez-Porras I, Canamero M, Rodriquez-Justo M, Serrano M, Barbacid M. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 19: 728-739, 2011. PMID: 21665147
- Guerra C, Schumacher A.J, Canamero M, Grippo P.J, Verdaguer L, Dubus P, Sandgren E.P, Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 11: 291-302, 2007. PMID: 17349585
- Harding H.P, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini D.D, Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol Cell 7: 1153-1163, 2001. PMID: 11430819
- Haruta I, Yanagisawa N, Kawamura S, Furukawa T, Shimizu K, Kato H, Koayashi M, Shiratori K, Yagi J. A mouse model of autoimmune pancreatitis with salivary gland involvement triggered by innate immunity via persistent exposure to avirulent bacteria. Lab Invest 90: 1757-1769, 2010. PMID: 20733561
- Heptner W, Neubauer H.P, Schleyerbach R. Glucose tolerance and insulin secretion in rabbits and dogs after ligation of the pancreatic ducts. Diabetologia 10: 193-196, 1974. PMID: 4602674
- Ida S, Ohmuraya M, Hirota M, Ozaki N, Hiramatsu S, Uehara H, Takamori H, Araki K, Baba H, Yamamura K. Chronic pancreatitis in mice by treatment with choline-deficient ethionine-supplemented diet. Exp Anim 59: 421-429, 2010. PMID: 20660988
- Iglesias A, Murga M, Laresgoiti U, Skoudy a, Bernales I, Fullaondo A, Moreno B, Lloreta J, Field S.J, Real F.X, Zubiaga A.M. Diabetes and exocrine pancreatic insufficiency in E2F1/E2F2 double-mutant mice. J Clin Invest 113: 1398-1407, 2004. PMID: 15146237
- Iida K, Li Y, McGrath B.C, Frank A, Cavener D.R. PERK eIF2 alpha kinase is required to regulate the viability of the exocrine pancreas in mice. BMC Cell Biol 8: 38, 2007. PMID: 17727724
- Ji B, Bi Y, Simeone D, Mortensen R.M, Logsdon C.D. Human pancreatic acinar cells lack functional responses to cholecystokinin and gastrin. Gastroenterology 121: 1380-1390, 2001. PMID: 11729117
- Ji B, Tsou L, Wang H, Gaiser S, Chang D.Z, Daniluk J, Bi Y, Longnecker D.S, Logsdon C.D. Ras activity levels control the development of pancreatic diseases. Gastroenterology 137: 1072-1082, 1082 e1-6, 2009. PMID: 19501586
- Jung I.H, Jung d.E, Park Y.N, Song S.Y, Park S.W. Aberrant Hedgehog ligands induce progressive pancreatic fibrosis by paracrine activation of myofibroblasts and ductular cells in transgenic zebrafish. PLoS One 6: e27941, 2011. PMID: 22164219
- Jupp J, D. Fine, C.D. Johnson. The epidemiology and socioeconomic impact of chronic pancreatitis. Best Pract Res Clin Gastroenterol 24: 219-231, 2010. PMID: 20510824
- Kayed H, Kleeff J, Keleg S, Buchler M.W, Friess H. Distribution of Indian hedgehog and its receptors patched and smoothened in human chronic pancreatitis. J Endocrinol 178: 467-478, 2003. PMID: 12967338
- Kanno H, Nose M, Itoh J, Taniquchi Y, Kyogoku M. Spontaneous development of pancreatitis in the MRL/Mp strain of mice in autoimmune mechanism. Clin Exp Immunol 89: 68-73, 1992. PMID: 1352748
- Lara L.F, M.J. Levy. Idiopathic recurrent acute pancreatitis. MedGenMed 6: 10, 2004. PMID: 15775837
- Li H.S, Zhang J.Y, Thompson B.S, Deng X.Y, Ford M.E, Wood P.G, Stolz D.B, Eagon P.K, Whitcomb D.C. Rat mitochondrial ATP synthase ATP5G3: cloning and upregulation in pancreas after chronic ethanol feeding. Physiol Genomics 6: 91-98, 2001. PMID: 11459924
- Lieber C.S, DeCarli L.M. The feeding of ethanol in liquid diets. Alcohol Clin Exp Res 10: 550-553, 1986. PMID: 3026198
- Li N, Wu X, Holzer R.G, Lee J.H, Todoric J, Park E.J, Ogata H, Gukovaskaya A.S, Gukovsky I, Pizzo D.P, VandenBerg S, Tarin D, Atay C, Arkan M.C, Deerinck T.J, Moscat J, Diaz-Meco M, Dawson D, Erkan M, Kleeff J, Karin M. Loss of acinar cell IKKalpha triggers spontaneous pancreatitis in mice. J Clin Invest 123: 2231-2243, 2013. PMID: 23563314
- Lohr M, Maisonneuve P, Lowenfels A.B. K-Ras mutations and benign pancreatic disease. Int J Pancreatol 27: 93-103, 2000. PMID: 10862508
- Lugea A, Tischler D, Nguyen J, Gong J, Gukovsky I, French S.W, Gorelick F.S, Pandol S.J. Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology 140: 987-997, 2011. PMID: 21111739
- Marrache F, Tu S.P, Bhagat G, Pendyala S, Osterreicher C.H, Gordon S, Ramanathan V, Penz-Osterreicher M, Betz K.S, Song Z, Wang T.C. Overexpression of interleukin-1beta in the murine pancreas results in chronic pancreatitis. Gastroenterology 135: 1277-1287, 2008. PMID: 18789941
- Masamune A, Kikuta K, Watanabe T, Satoh K, satoh A, Shimosegawa T. Pancreatic stellate cells express Toll-like receptors. J Gastroenterol 43: 352-362, 2008. PMID: 18592153
- Merkord J, Hennighausen G. Acute pancreatitis and bile duct lesions in rat induced by dibutyltin dichloride. Exp Pathol 36: 59-62, 1989. PMID: 2731591
- Merkord J, Jonas L, Weber H, Kroning G, Nizze H, Hennighausen G. Acute interstitial pancreatitis in rats induced by dibutyltin dichloride (DBTC): pathogenesis and natural course of lesions. Pancreas 15: 392-401, 1997. PMID: 9361094
- Merkord J, Weber H, Kroning G, Hennighausen G. Repeated administration of a mild acute toxic dose of di-n-butyltin dichloride at intervals of 3 weeks induces severe lesions in pancreas and liver of rats. Hum Exp Toxicol 20: 386-392, 2001. PMID: 11727788
- Miyahara T, Kawabuchi M, Goto M, Nakano I, Nada O, Nawata H. Morphological study of pancreatic endocrine in an experimental chronic pancreatitis with diabetes induced by stress and cerulein. Ultrastruct Pathol 23: 171-180, 1999. PMID: 10445284
- Mizunuma T, S. Kawamura, Y. Kishino. Effects of injecting excess arginine on rat pancreas. J Nutr 114: 467-471, 1984. PMID: 6199486
- Mori M, Fu X, Chen L, Zhang G, Higuchi K. Hereditary pancreatitis model WBN/Kob rat strain has a unique haplotype in the Pdwk1 region on chromosome 7. Exp Anim 58: 409-413, 2009. PMID: 19654439
- Mundlos S, Adler G, Schaar M, Koop I, Arnold R.. Exocrine pancreatic function in oleic acid-induced pancreatic insufficiency in rats. Pancreas 1: 29-36, 1986. PMID: 2437560
- Murphy J.A, Criddle D.N, Sherwood M, Chvanov M, Mukherjee R, McLaughlin E, Booth D, Gerasimenko J.V, Raraty M.G, Ghaneh P, Neoptolemos J.P, Gerasimenko O.V, Tepikin A.V, Green G.M, Reeve J.R Jr, Petersen O.H, Sutton R. Direct activation of cytosolic Ca2+ signaling and enzyme secretion by cholecystokinin in human pancreatic acinar cells. Gastroenterology 135: 632-641, 2008. PMID: 18555802
- Neuschwander-Tetri B.A, Burton F.R, Presti M.E, Britton R.S, Janney C.G, Garvin P.R, Brunt E.M, Galvin N.J, Poulos J.E. Repetitive self-limited acute pancreatitis induces pancreatic fibrogenesis in the mouse. Dig Dis Sci 45: 665-674, 2000. PMID: 10759232
- Niederau C, Luthen R, Niederau M.C, Grendell J.H, Ferrell L.D. Acute experimental hemorrhagic-necrotizing pancreatitis induced by feeding a choline-deficient, ethionine-supplemented diet. Methodology and standards. Eur Surg Res 24: 40-54, 1992. PMID: 1601023
- Ohashi K, Kim J.H, Hara H, Aso R, Akimoto T, Nakama K. WBN/Kob rats. A new spontaneously occurring model of chronic pancreatitis. Int J Pancreatol 6: 231-247, 1990. PMID: 1698893
- Ohashi S, Nishio A, Nakmura H, Kido M, Ueno S, Uza N, Inoue S, Kitamura H, Kiriya K, asada M, Tamaki H, Matsuura M, Kawasaki K, Fukui T, Watanabe N, Nakase H, Yodoi J, Okazaki K, Chiba T. Protective roles of redox-active protein thioredoxin-1 for severe acute pancreatitis. Am J Physiol Gastrointest Liver Physiol 290: G772-G781, 2006. PMID: 163222089
- Ohashi S, Nishio A, Nakamura H, Asada M, Tamaki H, Kawasaki K, Fukuio T, Yodoi J, Chiba T. Overexpression of redox-active protein thioredoxin-1 prevents development of chronic pancreatitis in mice. Antioxid Redox Signal 8: 1835-1845, 2006. PMID: 16987036
- Ohmuraya M, Hirota M, Araki K, Baba H, Yamamura K. Enhanced trypsin activity in pancreatic acinar cells deficient for serine protease inhibitor kazal type 3. Pancreas 33: 104-106, 2006. PMID: 16804421
- Ohmuraya M, Hirota M, Araki M, Mizushima N, Matsui M, Mizumoto T, Haruna K, Kume S, Takeya M, Ogawa M, Araki K, Yammura K. Autophagic cell death of pancreatic acinar cells in serine protease inhibitor Kazal type 3-deficient mice. Gastroenterology 129: 696-705, 2005. PMID: 16083722
- Ooi C.Y, Dorfman R, Cipolli M, Gonska T, Castellani C, Keenan K, Freedman S.D, Zielenski J, Berthiaume Y, Corey M, Schibli S, Tullis E, Durie P.R. Type of CFTR mutation determines risk of pancreatitis in patients with cystic fibrosis. Gastroenterology 140: 153-161, 2011. PMID: 20923678
- Ostrowski S.E, Reilly A.A, Collins D.N, Ramsingh A.L. Progression or resolution of coxsackievirus B4-induced pancreatitis: a genomic analysis. J Virol 78: 8229-8237, 2004. PMID: 15254194
- Perides G, Tao X, West N, Sharma A, Steer M.L. A mouse model of ethanol dependent pancreatic fibrosis. Gut 54: 1461-1467, 2005. PMID: 15870229
- Perides, G, Laukkarinen J.M, Vassileva G, Steer M.L. Biliary acute pancreatitis in mice is mediated by the G-protein-coupled cell surface bile acid receptor Gpbar1. Gastroenterology 138: 715-725, 2010. PMID: 19900448
- Pfutzer R.H, Barmada M.M, Brunskill A.P, Finch R, Hart P.S, Neoptolemos J, Furey W.F, Whitcomb D.C. SPINK1/PSTI polymorphisms act as disease modifiers in familial and idiopathic chronic pancreatitis. Gastroenterology 119: 615-623, 2000. PMID: 10982753
- Pfutzer, R.H, Tadic S.D, Thompson B.S, Zhang J.Y, Ford M.E, Eagon P.K, Whitcomb D.C. Pancreatic cholesterol esterase, ES-10, and fatty acid ethyl ester synthase III gene expression are increased in the pancreas and liver but not in the brain or heart with long-term ethanol feeding in rats. Pancreas 25: 101-106, 2002. PMID: 12131779
- Puig-Divi, V, Molero X, Salas A, Guarner F, Guarner L, Malagelada J.R. Induction of chronic pancreatic disease by trinitrobenzene sulfonic acid infusion into rat pancreatic ducts. Pancreas 13: 417-424, 1996. PMID: 8899803
- Qu W.M, Miyazaki T, Terada M, Okada K, Mori S, Kanno H, Nose M. A novel autoimmune pancreatitis model in MRL mice treated with polyinosinic:polycytidylic acid. Clin Exp Immunol 129: 27-34, 2002. PMID: 12100019
- Ramsingh A.I, Lee W.T, Collins D.N, Armstrong L.E. Differential recruitment of B and T cells in coxsackievirus B4-induced pancreatitis is influenced by a capsid protein. J Virol 71: 8690-8697, 1997. PMID: 9343227
- Rogers C.S, Stoltz D.A, Meyerolz D.K, Ostedgaard L.S, Rokhlina T, Taft P.J, Rogan M.P, Pezzulo A.A, Karp P.H, Itani O.A, Kabel A.C, Wohlford-Lenane C.L, Davis G.J, Hanfland R.A, Smith T.L, Samuel M, Wax D, Murphy C.N, Rieke A, Whitworth K, Uc A, Starner T.D, Brogden K.A, Shilyansky J, McCray P.B. Jr, Zabner J, Prather R.S, Welsh M.J. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 321: 1837-1841, 2008. PMID: 18818360
- Romac J.M, Ohmuraya M, Bittner C, Majeed M.F, Vigna S.R, Que J, Fee B.E, Wartmann T, Yamamura K, Liddle R.A. Transgenic expression of pancreatic secretory trypsin inhibitor-1 rescues SPINK3-deficient mice and restores a normal pancreatic phenotype. Am J Physiol Gastrointest Liver Physiol 298: G518-G524, 2010. PMID: 20110462
- Sah R.P, Dudeja V, Dawra R.K, Saluja A.K. Caerulein-Induced Chronic Pancreatitis Does Not Require Intra-Acinar Activation of Trypsinogen in Mice. Gastroenterology, 2013. PMID: 23354015
- Sarles H. Etiopathogenesis and definition of chronic pancreatitis. Dig Dis Sci 31: 91S-107S, 1986. PMID: 3525051
- Sarles H, Sahel J. Pathology of chronic calcifying pancreatitis. Am J Gastroenterol 66: 117-139, 1976. PMID: 788498
- Schmitz J.C, Protiva P, Gattu A.K, Utsumi T, Iwakiri Y, Neto A.G, Quinn M, Cornwell M.L, Fitchev P, Lugea A, Crawford S.E, Chung C. Pigment epithelium-derived factor regulates early pancreatic fibrotic responses and suppresses the profibrotic cytokine thrombospondin-1. Am J Pathol 179: 2990-2999, 2011. PMID: 21964188
- Scoggins C.R, Meszoely I.M, Wada M, Means A.L, Yang L, Leach S.D. p53-dependent acinar cell apoptosis triggers epithelial proliferation in duct-ligated murine pancreas. Am J Physiol Gastrointest Liver Physiol 279: G827-G836, 2000. PMID: 11005771
- Selig L, Sack U, Gaiser S, Kloppel G, Savkovic V, Mossner K, Keim V, Bodeker H. Characterisation of a transgenic mouse expressing R122H human cationic trypsinogen. BMC Gastroenterol 6: 30, 2006. PMID: 17069643
- Seok J, Warren H.S, Cuenca A.G, Mindrinos M.N, Baker H.V, Xu W, Richards D.R, McDonald-Smith G.P, Gao H, Hennessy L, Finnerty C.C, Lopez C.M, Honari S, Moore E.E, Minei J.P, Cuschieri K, Bakey P.E, Johnson J.L, Sperry J, Nathens A.B, Billiar T.R, West M.A, Jeschke M.G, Klein M.B, Gamelli R.L, Gibran N.S, Brownstein B.H, Miller-Graziano C, Calvano S.E, Mason P.H, Cobb J.P, Rahme L.G, Lowry S.F, Maier R.V, Moldawer L.L, Herdon D.N, Davis R.W, Xiao W, Tompkins R.G. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A 110: 3507-3512, 2013. PMID: 23401516
- Snouwaert J.N, Brigman K.K, Latour A.M, Malouf N.N, Boucher R.C, Smithies O, Koller B.H. An animal model for cystic fibrosis made by gene targeting. Science 257: 1083-1088, 1992. PMID: 1380723
- Sparmann, G, Merkord J, Jaschke a, Nizze HH, Jonas L, Lohr M, Liebe S, Emmrich J. Pancreatic fibrosis in experimental pancreatitis induced by dibutyltin dichloride. Gastroenterology 112: 1664-1672, 1997. PMID: 9136846
- Sun X, Sui H, Fisher J.T, Yan Z, Liu X, Cho H.J, Joo N.S, Zhang Y, Zhou W, Yi Y, Kinyon J.M, Lei-Butters D.C, Griffin M.A, Naumann P, Luo M, Ascher J, Wang K, Frana T, Wine J.J, Meyerholz D.K, Engelhardt J.F. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J Clin Invest 120: 3149-3160, 2010. PMID: 20739752
- Swanson G, Forsyth C.B, Tang Y, Shaikh M, Zhang L, Turek F.W, Keshavarzian A. Role of intestinal circadian genes in alcohol-induced gut leakiness. Alcohol Clin Exp Res 35: 1305-1314, 2011. PMID: 21463335
- Tanaka T, Ichiba Y, Fujoo Y, Itoh H, Kodama O, Dohi K. New canine model of chronic pancreatitis due to chronic ischemia with incomplete pancreatic duct obstruction. Digestion 41: 149-155, 1988. PMID: 3224767
- Treiber M, Schulz H.U, Landt O, Drenth J.P, Castellani C, Real F.X, Akar N, Ammann R.W, Bargetzi M, Bhatia E, Demaine A.G, Battagia C, Kingsnorth A, O'Reilly d, Truninger K, Koudova M, Spicak J, Cerny M, Menzel H.J, Moral P, Pignatti P.F, Romanelli M.G, Rickards O, De Stefano G.F, zarnescu N.O, Choudhuri G, sikora S.S, Jansen J.B, Weiss F.U, Pietschmann M, Teich N, Gress T.M, Ockenga J, Schmidt H, Kage A, Halangk J, Rosendahl J, Gronenberg D.A, Nickel R, Witt H. Keratin 8 sequence variants in patients with pancreatitis and pancreatic cancer. J Mol Med (Berl) 84: 1015-1022, 2006. PMID: 17039343
- Tsukamoto H, Sankaran H, Delgado G, Reidelberger R.D, Deveney C.W, Largman C. Increased pancreatic acinar content and secretion of cationic trypsinogen following 30-day continuous ethanol intoxication in rats. Biochem Pharmacol 35: 3623-3629, 1986. PMID: 3768045
- Uc A, Giriyappa R, Meyerholz D.K, Griffin M, Ostedgaard L.S, Tang X.X, Abu-El-Haija M, Stoltz D.A, Ludwig P, Pezzulo A, Taft P, Welsh M.J. Pancreatic and biliary secretion are both altered in cystic fibrosis pigs. Am J Physiol Gastrointest Liver Physiol 303: G961-G968, 2012. PMID: 22936270
- Uchida K, Okazaki K, Nishi T, Uose S, Nakase H, Ohana M, Matsushima Y, Omori K, Chiba T. Experimental immune-mediated pancreatitis in neonatally thymectomized mice immunized with carbonic anhydrase II and lactoferrin. Lab Invest 82: 411-424, 2002. PMID: 11950899
- Vardanyan M, Melemedjian O.K, Price T.J, Ossipov M.H, Lai J, Roberts E, Boos T.L, Deschamps J.R, Jacobson A.E, Rice K.C, Porreca F. Reversal of pancreatitis-induced pain by an orally available, small molecule interleukin-6 receptor antagonist. Pain 151: 257-265, 2010. PMID: 20599324
- Vaquero E,Molero X, Tian X, Salas A, Malagelada J.R. Myofibroblast proliferation, fibrosis, and defective pancreatic repair induced by cyclosporin in rats. Gut 45: 269-277, 1999. PMID: 10403741
- Vonlaufen A, Phillips P.A, Xu Z, Zhang X, Yang L, Pirola R.C, Wilson J.S, Apte M.V. Withdrawal of alcohol promotes regression while continued alcohol intake promotes persistence of LPS-induced pancreatic injury in alcohol-fed rats. Gut 60: 238-246, 2011. PMID: 20870739
- Vonlaufen A, Xu Z, Daniel B, Kumar R.K, Pirola R, Wilson J, Apte M.V. Bacterial endotoxin: a trigger factor for alcoholic pancreatitis? Evidence from a novel, physiologically relevant animal model. Gastroenterology 133: 1293-1303, 2007. PMID: 17919500
- Watanabe S, Abe K, Anbo Y, Katoh H. Changes in the mouse exocrine pancreas after pancreatic duct ligation: a qualitative and quantitative histological study. Arch Histol Cytol 58: 365-374, 1995. PMID: 8527243
- Weaver C, A.E. Bishop, J.M. Polak. Pancreatic changes elicited by chronic administration of excess L-arginine. Exp Mol Pathol 60: 71-87, 1994. PMID: 8070543
- Weiss F.U, Simon P, Bogdanova N, Mayerle J, Dworniczak B, Horst J, Lerch M.M. Complete cystic fibrosis transmembrane conductance regulator gene sequencing in patients with idiopathic chronic pancreatitis and controls. Gut 54: 1456-1460, 2005. PMID: 15987793
- Whitcomb D.C. Value of genetic testing in the management of pancreatitis. Gut 53: 1710-1717, 2004. PMID: 15479696
- Whitcomb, D.C. Hereditary pancreatitis: new insights into acute and chronic pancreatitis. Gut 45: 317-322, 1999. PMID: 10446089
- Whitcomb D.C, Gorry M.C, Preston R.A, Furey W, Sossenheimer M.J, Ulrich C.D, Martin S.P, Gates L.K Jr, Amann S.T, Toskes P.P, Liddle R, McGrath K, Uomo G, Post J.C, Ehrlich G.D. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet 14: 141-145, 1996. PMID: 8841182
- Winston J.H, He Z.J, Shenoy M, Xiao S.Y, Pasricha P.J. Molecular and behavioral changes in nociception in a novel rat model of chronic pancreatitis for the study of pain. Pain 117: 214-222, 2005. PMID: 16098667
- Wilson J.S, Apte M.V. Role of alcohol metabolism in alcoholic pancreatitis. Pancreas 27: 311-315, 2003. PMID: 14576493
- Witt H, Apte M.V, Keim V, Wilson J.S. Chronic pancreatitis: challenges and advances in pathogenesis, genetics, diagnosis, and therapy. Gastroenterology 132: 1557-1573, 2007. PMID: 17466744
- Yadav D, Timmons L, Benson J.T, Dierkhising R.A, Chari S.T. Incidence, prevalence, and survival of chronic pancreatitis: a population-based study. Am J Gastroenterol 106: 2192-2199, 2011. PMID: 21946280
- Yamamoto M, Otani M, Otsuki M. A new model of chronic pancreatitis in rats. Am J Physiol Gastrointest Liver Physiol 291: G700-G708, 2006. PMID: 16959955
- Yamaguchi T, Kihara Y, Taquchi M, Naqashio Y, Tashiro M, Nakamura H, Otsuki M. Persistent destruction of the basement membrane of the pancreatic duct contributes to progressive acinar atrophy in rats with experimentally induced pancreatitis. Pancreas 31: 365-372, 2005. PMID: 16258372
- Yamashina M, Nishio A, Nakayama S, Okazaki T, Uchida K, Fukui T, Okazaki K. Comparative study on experimental autoimmune pancreatitis and its extrapancreatic involvement in mice. Pancreas 41: 1255-1262, 2012. PMID: 22836854
- Zhu Y, Colak T, Shenoy M, Liu L, Pai R, Li C, Mehta K, Pasricha P.J. Nerve growth factor modulates TRPV1 expression and function and mediates pain in chronic pancreatitis. Gastroenterology 141: 370-377, 2011. PMID: 21473865
- Zhu Y, Mehta K, Li C, Xu G.Y, Liu L, Colak T, shenoy M, Pasricha P.J. Systemic administration of anti-NGF increases A-type potassium currents and decreases pancreatic nociceptor excitability in a rat model of chronic pancreatitis. Am J Physiol Gastrointest Liver Physiol 302: G176-G181, 2012. PMID: 22038828