
Methods Type:
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2021.13
| Attachment | Size |
|---|---|
| 1.04 MB |
Since the introduction of the first genetically engineered mouse models (GEMM) based on oncogene expression of Myc and TGFαvia the elastase promoter, GEMMs have provided an invaluable tool for experimentally targeting tumor biological, microenvironmental and translational questions (20, 29, 40, 70, 80, 100). The characterization of pancreatic carcinogenesis based on Kras activation and step-wise loss of tumor suppressor genes including p16ink4a, p53 and SMAD4 and the identification of PanIN as relevant precursor lesion in the decade of the 1990s, allowed the development of KRASG12D-based mouse models that inherently recapitulated the paradigm observed in human pancreatic ductal adenocarcinoma (PDAC) (58). Over the past decade, a plethora of GEMMs combining the original KRASG12D-based model with additional loss-of-function alleles have helped understanding the role of high-frequent driver genes and molecular pathways during pancreatic tumor progression (70). Emerging fields of interest addressed by GEMMS include early tumor detection, response to therapy and screening for key biomarkers, and mechanisms of drug resistance (70). In the era of next-generation sequencing (NGS), a large catalogue of point mutations, indels and complex epigenetic alterations have been identified in PCa (70, 92). Genomic screening in mice using transposon-based insertional mutagenesis and CRISPR/Cas9 multiplexing have arisen as fundamental tools for identifying tumor-causing mutations, for dissecting the contribution of individual signaling pathways in PCa and aid the interpretation of large datasets resulting from NGS (67, 92).
I. KRAS-based genetically engineered mouse models of PDAC
The development of GEMMs of PDAC offers the possibility to reproduce the complexity of pancreatic tumorigenesis in controlled experimental systems (30) (Figure 1). In these models, pancreas-specific activation of a mutant KRAS oncogene during embryonic development triggers preneoplastic pancreatic intraepithelial lesions (PanINs), which can readily progress to invasive PDAC after concomitant inactivation of various tumor suppressor genes (29). The most characterized mutation during early pancreatic tumorigenesis is the activation of the KRAS oncogene, found in 74% of low-grade PanIN lesions and over 90% of invasive tumors (29, 40). In human PCa, the most frequently altered tumor suppressor gene is CDKN2A gene, found inactivated, deleted, or epigenetically silenced in 95% of pancreatic tumors (29). Additional acquired genetic alterations during PanIN progression are often associated to inactivation of the tumor suppressor genes Trp53 and SMAD4 present in 75% of the cases with progression of PanIN-3 lesions to invasive PDAC tumors (29, 70).
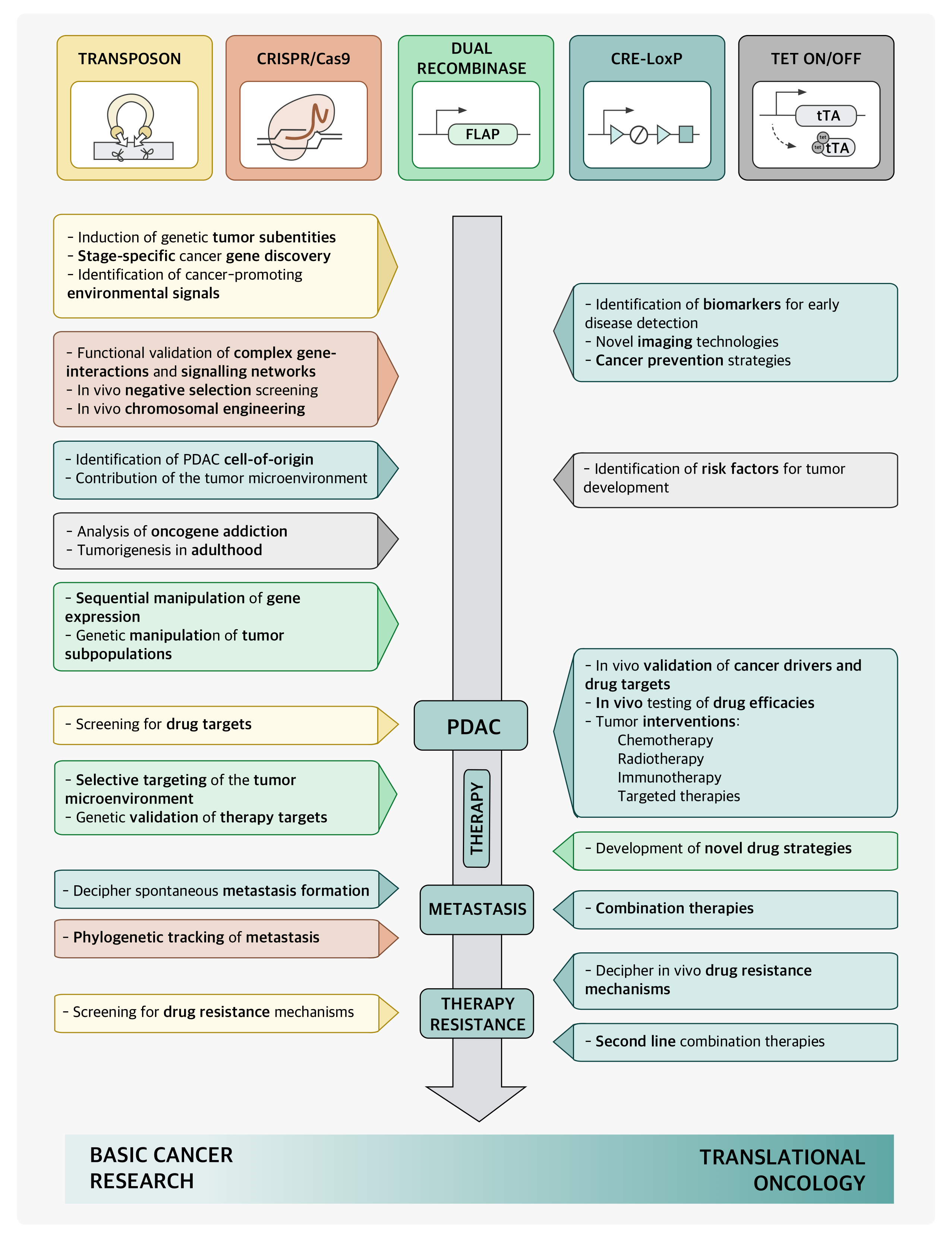
Figure 1. Generation of mutant Kras-driven GEMMs of PDAC based on the Cre/loxP-recombination system. First, Cre/loxP-mediated conditional activation/inactivation of oncogenes or tumor suppressor genes in the pancreas directed by the Pdx1-promotor. Second, GEMM reproduce tumors that recapitulate different types of human preneoplastic lesions and PDAC depending on the inactivated tumor suppressor gene. TSG, tumor suppressor genes; PanIN, pancreatic intraepithelial neoplasia; IPMN, intraductal papillary mucinous neoplasia; MCN, mucinous neoplasia.
Table 1. Prenatal GEMMs of Pancreas Cancer.
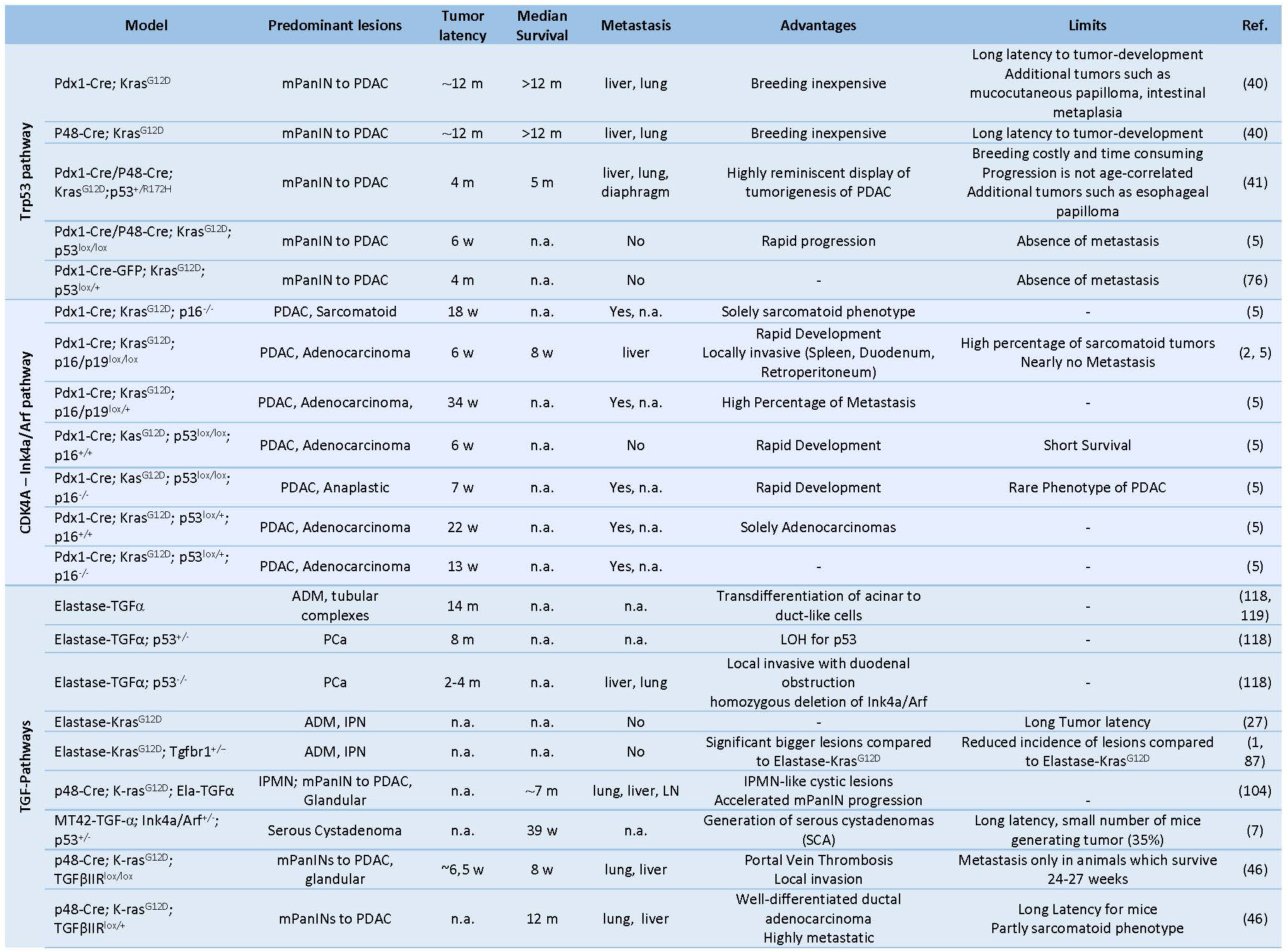
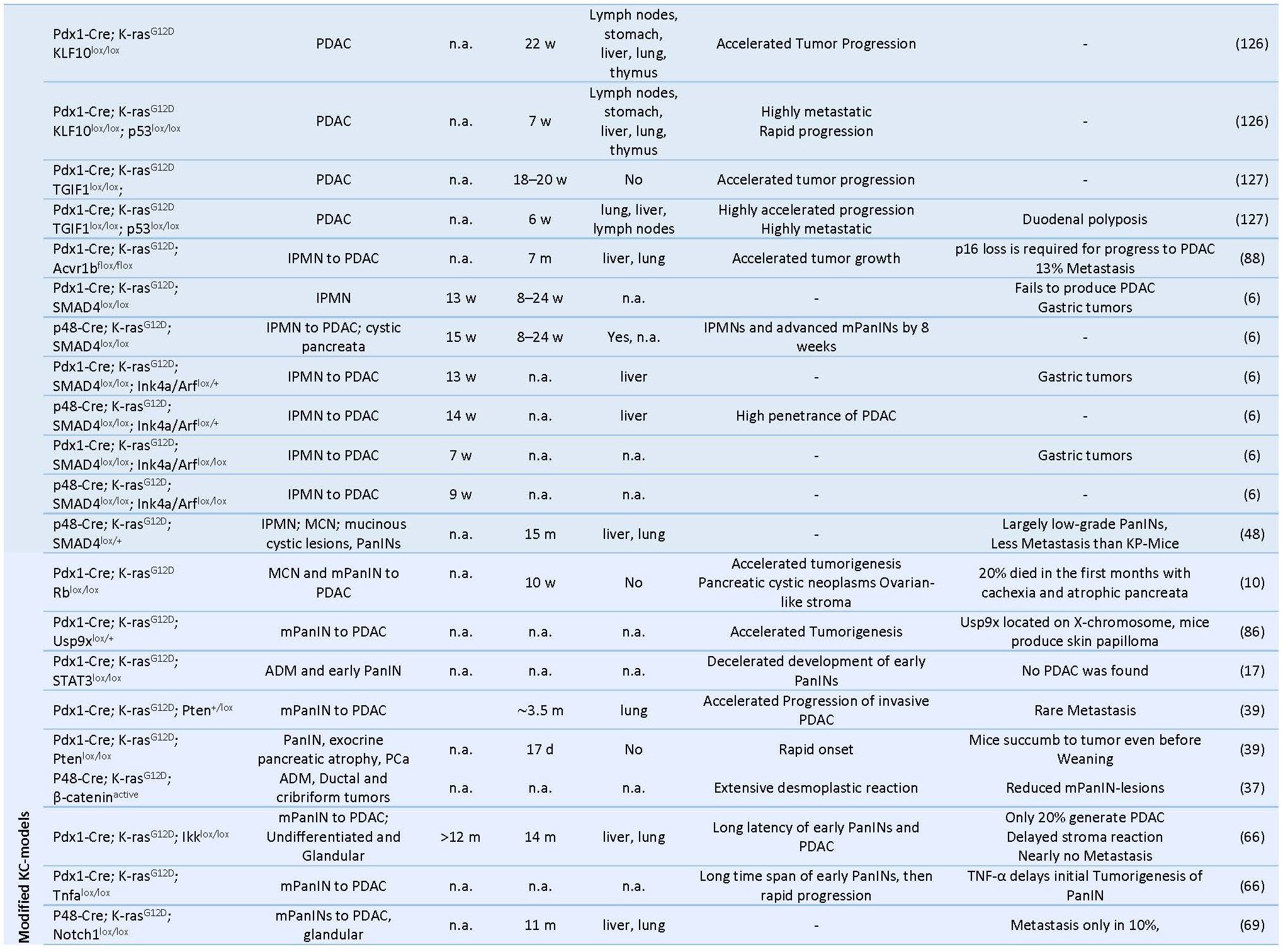
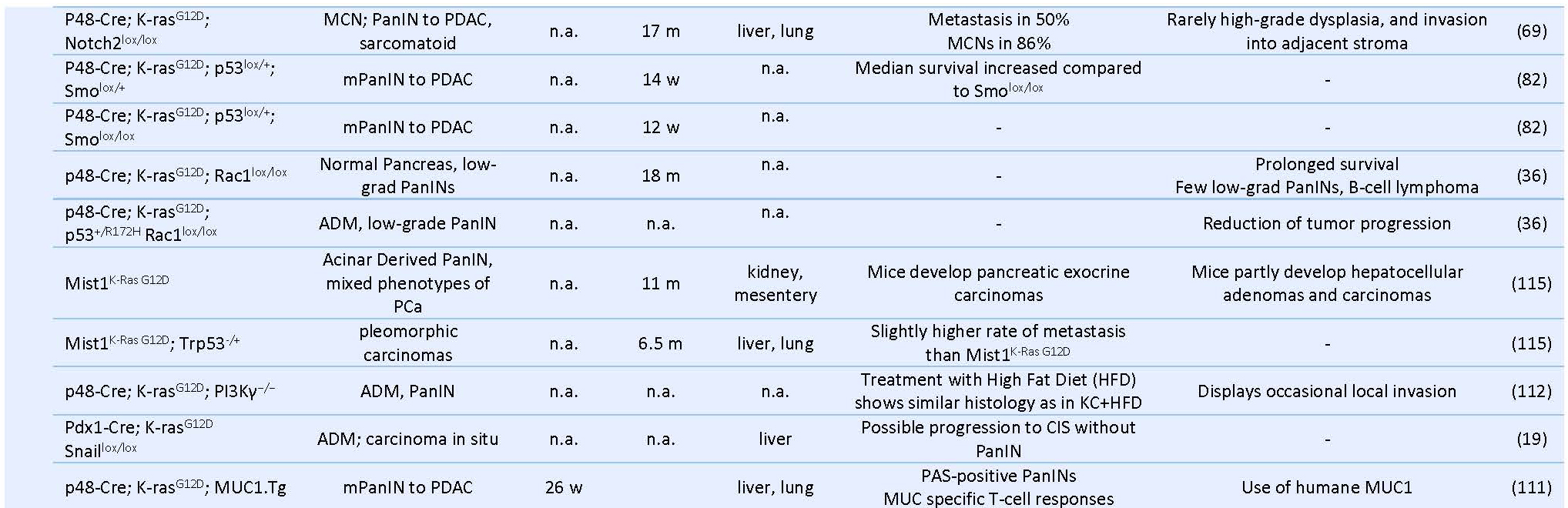 PDAC: pancreatic ductal adenocarcinoma, PCa: pancreatic cancer, mPanIN: murine pancreatic intraepithelial neoplasia, IPMN: intraductal papillary mucinous neoplasm, MCN: mucinous cystic neoplasm, IPN: intra ductal papillary neoplasms, CPN: cystic papillary neoplasm, ADM: acinar-to-ductal metaplasia, LOH: Loss of heterozygosity, CIS: carcinoma in situ, m: months, w: weeks, n.a.: not available.
PDAC: pancreatic ductal adenocarcinoma, PCa: pancreatic cancer, mPanIN: murine pancreatic intraepithelial neoplasia, IPMN: intraductal papillary mucinous neoplasm, MCN: mucinous cystic neoplasm, IPN: intra ductal papillary neoplasms, CPN: cystic papillary neoplasm, ADM: acinar-to-ductal metaplasia, LOH: Loss of heterozygosity, CIS: carcinoma in situ, m: months, w: weeks, n.a.: not available.The first successful GEMM of PCa termed “KC” mice involved a conditional Cre/loxP-based activation of an endogenous knocked-in KRASG12D oncogene in the pancreatic lineage leading to the activation of downstream proteins like ERK and c-myc and increased mitogen activity (29, 40). Here, mice carrying a mutant KRASLSL-G12Dknock-in allele silenced by the insertion of a LoxP-flanked transcription/translation STOP cassette was crossed to Pdx1 or Ptf1a/p48 transgenic strains expressing Cre-recombinase targeting pancreatic progenitor cells (31, 40) (Table 1) (Figure 2). The pancreata of compound mutant mice developed, with complete penetrance, the full spectrum of human PanIN lesions, pinpointing KRASG12D mutation as the initiating event in PCa (40). Consistent with its cognate human condition, invasive tumors developed after a long latency at advanced age of the mice (12-15 months) (70). The use of KC mice for translational therapeutic approaches is strictly limited by the late and barely predictable development of PDAC (70). In order to accelerate tumor progression to invasive PDAC, a variety of GEMMs has been developed over the past decade by introducing additional genetic alterations in loci encoding tumor suppressor genes known to be commonly mutated in human PCa such as CDKN2A/p16Ink4a, TP53, BRCA2 or SMAD4 (29) (Table 1) (Figure 3). Activating point mutations in the KRAS oncogene along with CDKN2A/p16Ink4a inactivation and altered expression of TGF-β are almost universal events in human PCa (5, 50). Homozygous deletion of the Ink4a/Arf locus in the KC model generated by interbreeding KRASG12D, p16fl/fl and Pdx1-Cre mice, results in a greatly curtailed median survival of 8 weeks due to accelerated PanIN progression, a reduced tumor latency with locally invasive and poorly differentiated tumor development, which showed microscopic metastasis to liver and lung (41, 70). Similarly, mice with homozygous loss or dominant-negative mutations in the Trp53 tumor suppressor gene, named thereafter KPC mice, closely resemble human PCa as they developed PanIN lesions by the age of 8 weeks and PDAC with an invasive and well-differentiated phenotype by the age of 16 weeks (35). Interestingly, there seem to be some differences between activation of a dominant-negative p53R172H mutation and biallelic conditional loss of p53, including a more rapid development of invasive PDAC with decreased survival of the Trp53-/- mice and the lack of the highly metastatic phenotype characteristic of mice with mutant p53R172H (41, 70). Also, the engineered homozygous loss of the p53 allele leads to the formation of a highly undifferentiated anaplastic tumor, whereas engineered heterozygous loss of p53 leads to progression to PDAC with kinetics similar to the R172H mutant mice.
Combined GEMMs with inactivation of SMAD4 or transcription intermediary factor 1 (TIF-1), an integral component of the transforming growth factor β pathway, exhibit early rapid development of cystic lesions within the ductal epithelium resembling to human intraductal papillary mucinous neoplasia (IPMN) and mucinous cystic neoplasia (71), yet failed to develop aggressive pancreatic neoplasms (35, 41, 54, 70). Furthermore, conditional deletion of Notch2 in KC mice abrogates its characteristic PanIN development and induces MCN-like lesions (70) (Table 1).
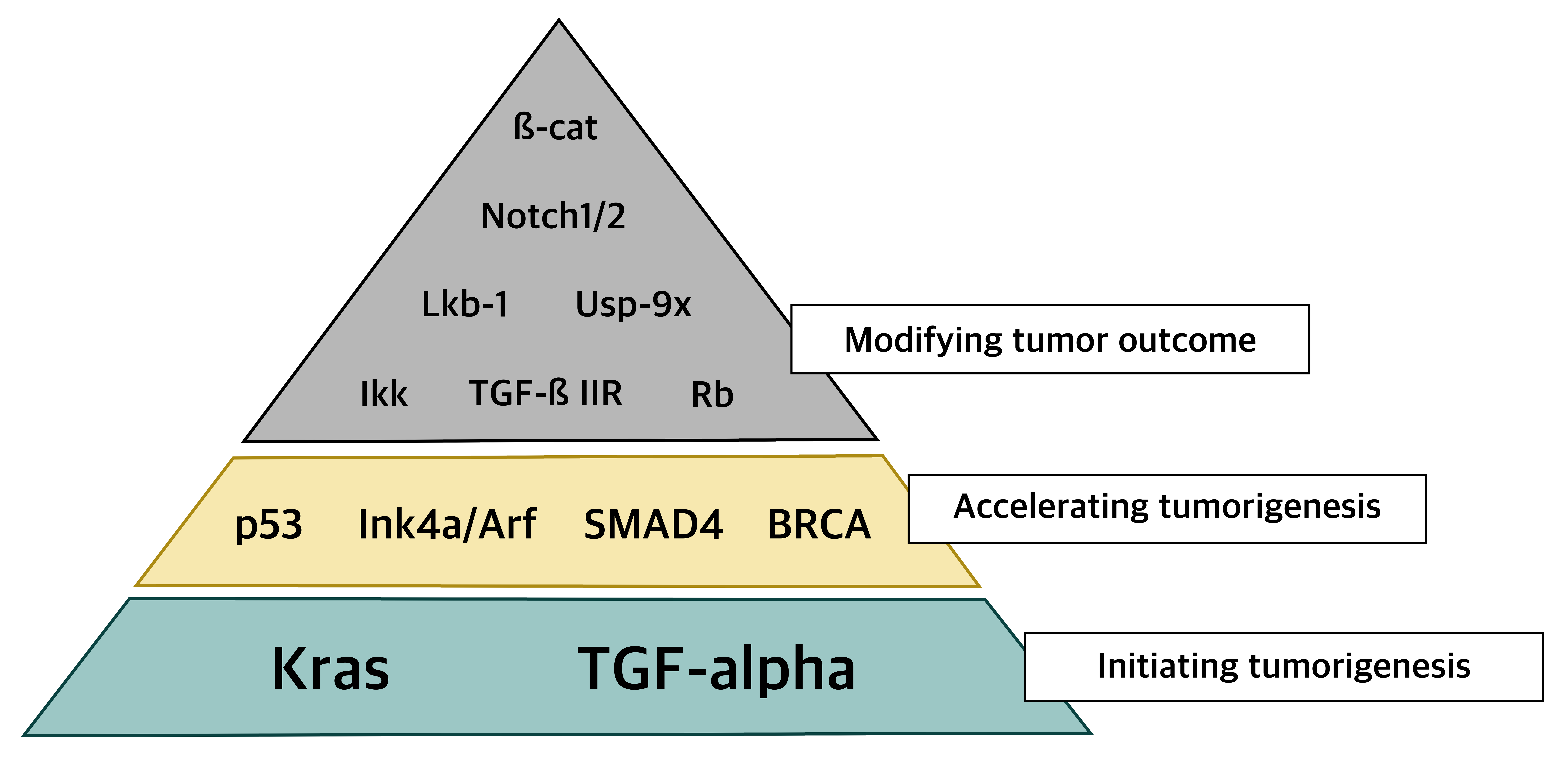
Figure 2. Drivers of genetic alterations in human pancreas cancer used for the generation of GEMMs of PDAC.
Hereditary predisposition accounts for around 10% of human PCa and mutations in BRCA2 locus constitute the most frequently encountered germline genetic alterations (105). Skoulidis et al. generated a mouse model for familial PCa by crossing the KPC strain with mice carrying a germline-truncated Brca2Tr allele and a floxed Brca2 allele, termed KPCBTr/fl11 mice (29, 105). Homozygous Brca2 inactivation in these mice caused PDAC tumors with higher penetrance and shorter latency than siblings carrying wild-type Brca2 alleles (median PDAC-free survival 84 days vs 168 days, respectively) (105) (Table2). Interestingly, germline heterozygosity for the truncating allele Brca2Tr also curtailed tumor latency in KPCBTr/WT mice, irrespective of the functional status of Trp53, indicating that loss of heterozygosity (43) is not an essential requirement for pancreatic tumor development (105). In another setting, addition of a conditional knock-out allele of LKB1 to the KC mouse model synergizes with activated KRAS and resulted in accelerated tumor development with reduced latency and likewise, without detectable LOH (29, 75) (Table 2).
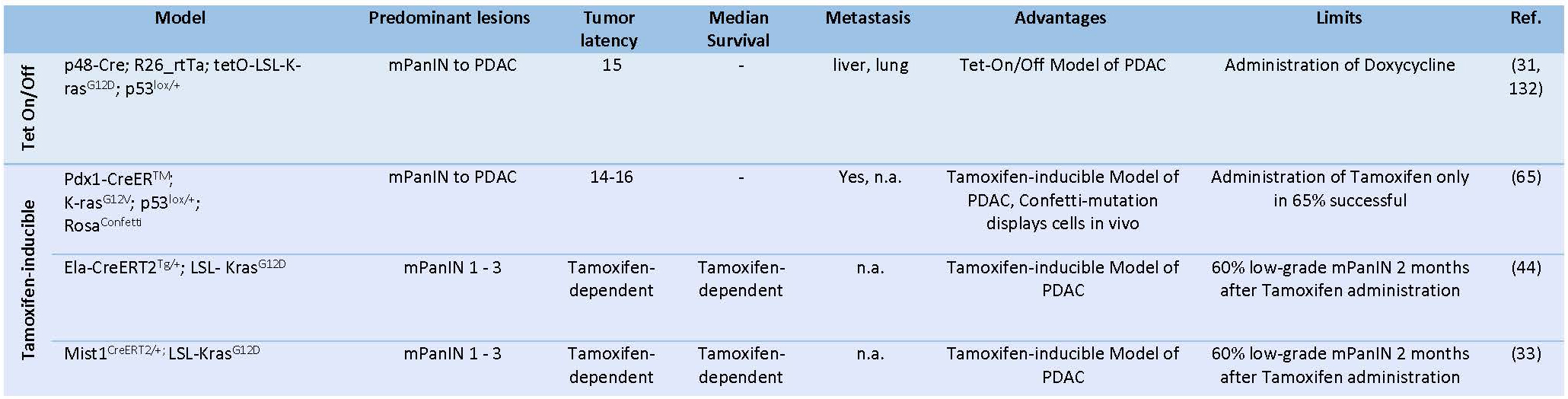
Despite remarkable similarities between human PCa and the pancreatic lesions observed in these GEMMs, their etiology is distinct from that of the human disease (29, 70). In contrast to the described models, human PDAC originates from somatic mutations in the KRAS oncogene during adulthood rather than during embryonic development and in selected cell types not in the entire pancreas (29, 30). To overcome some of these limitations, another model in which the expression of the oncogenic KRAS is restricted to acinar cells was generated by crossing a conditional KRASLSLG12Vgeo knock-in strain with double transgenic Elas-tTA/tetO-Cre mice (31). These mice express Cre-recombinase specifically in acinar cells under the control of the Elastase promoter following an inducible Tet-Off strategy (29, 31) (Table 3). This strategy allows to control the temporal expression of targeted KRASG12Voncogene by simply removing doxycycline from the drinking water of these animals (29, 31), which offers the possibility to evaluate the effect of KRAS oncogene during postnatal development (31). Unexpectedly, turning on the KRASG12V expression in acinar cells of the pancreata of adult mice failed to induce detectable lesions including metaplasia or low-grade PanINs, even in the presence of inactivated Trp53 or Ink4a/Arf tumor suppressors (29-31). However, these acinar cells, provided they harbour KRAS oncogene, yielded PanINs and PDAC upon induction of repetitive bouts of pancreatitis (29-31). As expected, the concomitant loss of the tumor suppressor genes Trp53 or Ink4a/Arf and cerulein-induced pancreatitis in KRASG12V background increased the penetrance of tumor development and enhanced its metastatic potential (29-31). These observations highlight that the multistep transformation process mediated by the activation of KRAS oncogene and loss of Trp53 during embryonic development can be reproduced in adult mice through the induction of chronic pancreatitis and the resulting temporary tissue damage(30).
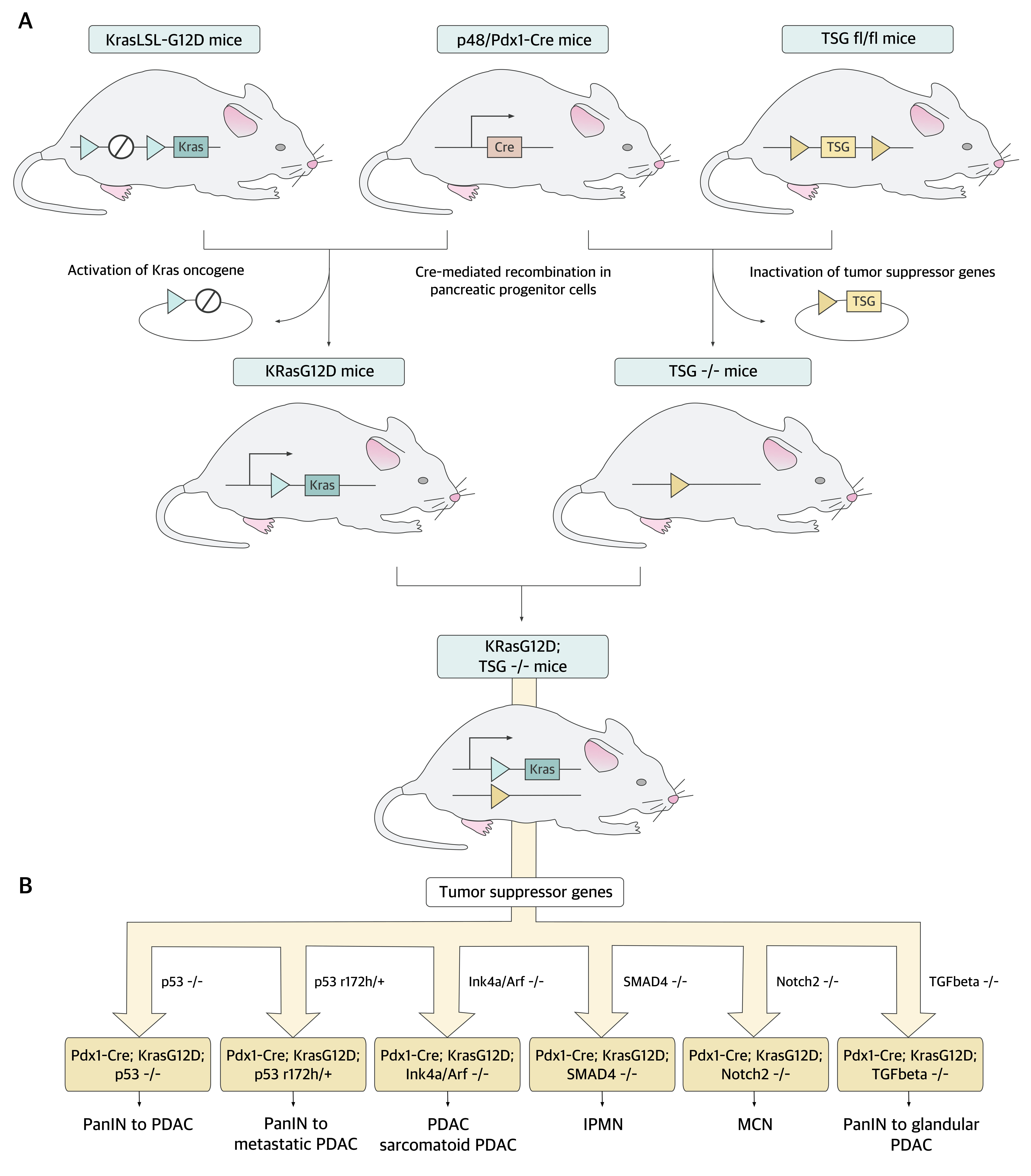
Figure 3. Application of transgenic PDAC mouse models in basic cancer research and translational oncology.
Table 2. GEMMs of hereditary PCa.

Table 3. Postnatal GEMMs of PCa.
II. TGFα-based transgenic mice
The overexpression of the epidermal growth factor receptor (EFGR) and its ligand, transforming growth factor-α (TGFα), constitute an additional key signaling pathway in human PDAC (119). The current generation of TGFαtransgenic mice are based on the Ela-TGFα strain generated by Sandgren et al, in which the activation of EGFR signaling was induced by acinar-specific transgenic expression of TGFα under the control of the rat elastase-1 promoter (101, 119). For the concomitant deletion of Trp53 and pancreatic overexpression of TGFα, these mice were interbred with Trp53fl/fl and Ptf1a-Creex1/+ mice generating the Ela-TGFα;Trp53Δ/Δ(TPC) mice (21). These mice demonstrate in ca. 66% of cases undifferentiated pancreatic tumors and in approximately 25% of cases PDAC with reduced median survival time compared to Ela-TGFα mice (300 vs 445 days, respectively) (21). Additional loss of the NF-kappaB/p65 signalling via conditional biallelic deletion of the RelA/p65 locus led to the emergence of well-differentiated tumors presenting in 75% of the cases a ductal phenotype (21). Here, Ela-TGFα mice were crossed with Trp53fl/fl mice, Ptf1a-Creex1/+ mice and RelA/p65fl/fl mice to generate Ela-TGFα;Trp53Δ/Δ;p65Δ/Δ (TPAC) mice (59). Unexpectedly, TPAC mice presented pancreatic tumors with human-like neural invasion, a pathognomonic feature of human PDAC, which has been precluded in all known GEMMs of PDAC to date (21)
III. Conditional insertional mutagenesis: a novel approach for cancer gene discovery
Transposon-based insertional mutagenesis provides a high-throughput non-biased platform for functional genomic screening in cancer animal models (91, 92). Endogenous DNA-transposons are genetic elements encoding a transposase protein with the ability to change their position within a genome by a cut-and-paste mechanism (16, 124). Sleeping Beauty (SB), a member of the Tc1/mariner class of transposons from fish, was the first transposon engineered to be active for somatic mutagenesis in mice (60, 92). Another transposon, piggyBac, originates from the cabbage looper moth and has been shown to be also functional in mammalian cells. Compared to SB, piggyBac exhibits several advantages including a higher transposition efficiency with mobilization of larger cargo segments, low frequencies of local hopping, no evidence of footprint mutation after transposition and a high tendency towards reintegration in intragenic regions (60) (Figure 4). In order to fully characterize pancreatic tumorigenesis, Rad et al. developed a transposon system with an oncogenic transposon that can be mobilized by both SB or piggyBac transposases (92). The duality of the generated transposon mouse lines enables optimal use of these two non-redundant complementary systems and facilitates saturation mutagenesis as observed in studies in D. melanogaster (92). To confine transposition to the pancreatic tissue, a piggyBac or SB transposase allele preceded by a loxP-flanked stop cassete (LSL) was knocked into the Rosa26 locus, which encodes an ubiquitously expressed non-essential gene and used it to generate Rosa26LSL-PB mice (52, 92). The expression of the transposase knock-in, which is normally silenced due to the flanking LSL-cassette, can be specifically activated in pancreatic tissue using the Pdx1-Cre knockin construct (52). For activation of transposition, Pdx1-cre; Rosa26LSL-PB were crossed with mutagenic activating transposon mice lines (Figure 4) (92). This highly versatile triple-transgenic mice model permits conditional mobilization of piggyBac transposase allele by Cre-recombinase to induce cancer in any tissue of interest (52). The incorporation of SB or piggyBac transposon systems in a KRASG12D background led to accelerated pancreatic tumor development with increased penetrance, shorter latency and substantially reduced survival independently of the number of copies of the oncogenic transposon (92). The resulting tumors were usually large, cystic and frequently metastasized (92).
Transposon-based technology enables the identification of genomic regions that are hit by transposons more frequently than predicted by chance, termed common insertion sites (123), which are therefore more likely to harbor cancer-causing genes (Figure 4) (52). The conditional piggyBac system bears a wide range of applications, including the analysis of stage-specific genetic events driving different phases of pancreatic tumorigenesis as well as the screening for therapeutic targets or drug resistance mechanisms (Figure 1) (92). The possibility to induce cancer subtypes or to reprogram differentiated somatic cells into therapeutically applicable pluripotent stem cells, lays the basis for further in-depth molecular studies (92, 133).
IV. Next generation dual-recombinase systems
Conventional Cre-loxP mouse models have revolutionized the understanding of pancreatic tumorigenesis and therapy resistance but are limited by their inability to recapitulate the multistep carcinogenesis and tumor heterogeneity as it is present in human PDAC (102). To tackle these problems, Schönhuber et al. developed an inducible dual-recombinase system, in which the universal Cre-LoxP technology was combined with a flippase-FRT (Flp-FRT)-recombination system directed by the mouse Pdx1 promoter (Pdx1-Flp) (102). In this model, oncogenic KRAS was conditionally activated in the Pdx1-Flp lineage of the Pdx1-Flp; FSF-KRASG12D/+ (79) mice containing a FSF-silenced KRAS knock-in allele (102). The sequential genetic manipulation is achieved by the introduction of a latent tamoxifen-inducible CreERT2allele silenced by an FRT-stop-FRT (FSF) cassette under the control of the CAG promoter as a Rosa26 knock in (FSF-R26CAG−CreERT2/+)(Figure 5) (102). These mice presented comparable tumor latency and survival rates to the widely-used KC model showing a human-like PDAC phenotype with minimal extrapancreatic disease (102).

Figure 4. Conditional piggyBac transposon system for genetic screening in murine PDAC. (A) To obtain tissue specific mutagenesis in the pancreas, mouse strains harboring transposon (ATP mouse lines) and transposase (Rosa26 miceLSL-PB) were crossed with KrasLSL-G12D floxed mice and a Cre driver line containing the pancreas-specific promoter Pdx1. (B) PiggyBac conditional transposition systems is designed to disrupt gene expression. Transposons contain in both ends recognition sites necessary for transposon activation by the transposase (purple spheres). After transposon excision, the remaining DNA is repaired by non-homologous end joining (NHEJ), while the mobilized transposon integrates into a TA-dinucleotide site within the parent genome. Based on the location and orientation of insertion, the transposon mediates either oncogenic activation or tumor suppressor disruption. (C) Functional differences of Sleeping Beauty and piggyBac transposases. (D) Experimental pipeline for the detection of common insertion sites in pancreas cancer.
The dual recombination systems enable to uncouple temporal activation of oncogenic KRAS from the inactivation of tumor suppressor genes such as Trp53 (102). The application of high-dose tamoxifen in 2-month-old mediated the activation of CreERT2 in Pdx1-Flp; FSF-KRASG12D/+; FSF-R26CAG−CreERT2/+ mice with floxed Trp53 leading to stage-specific elimination of p53 and the rapid generation of multifocal PDAC (102). This novel approach provides researchers with a powerful tool for genetic modelling of tumor subpopulations, selective targeting of noncancerous cell types in the tumor microenvironment, and genetic validation of therapeutic targets on a genome-wide scale in vivo (Figure 1) (102).
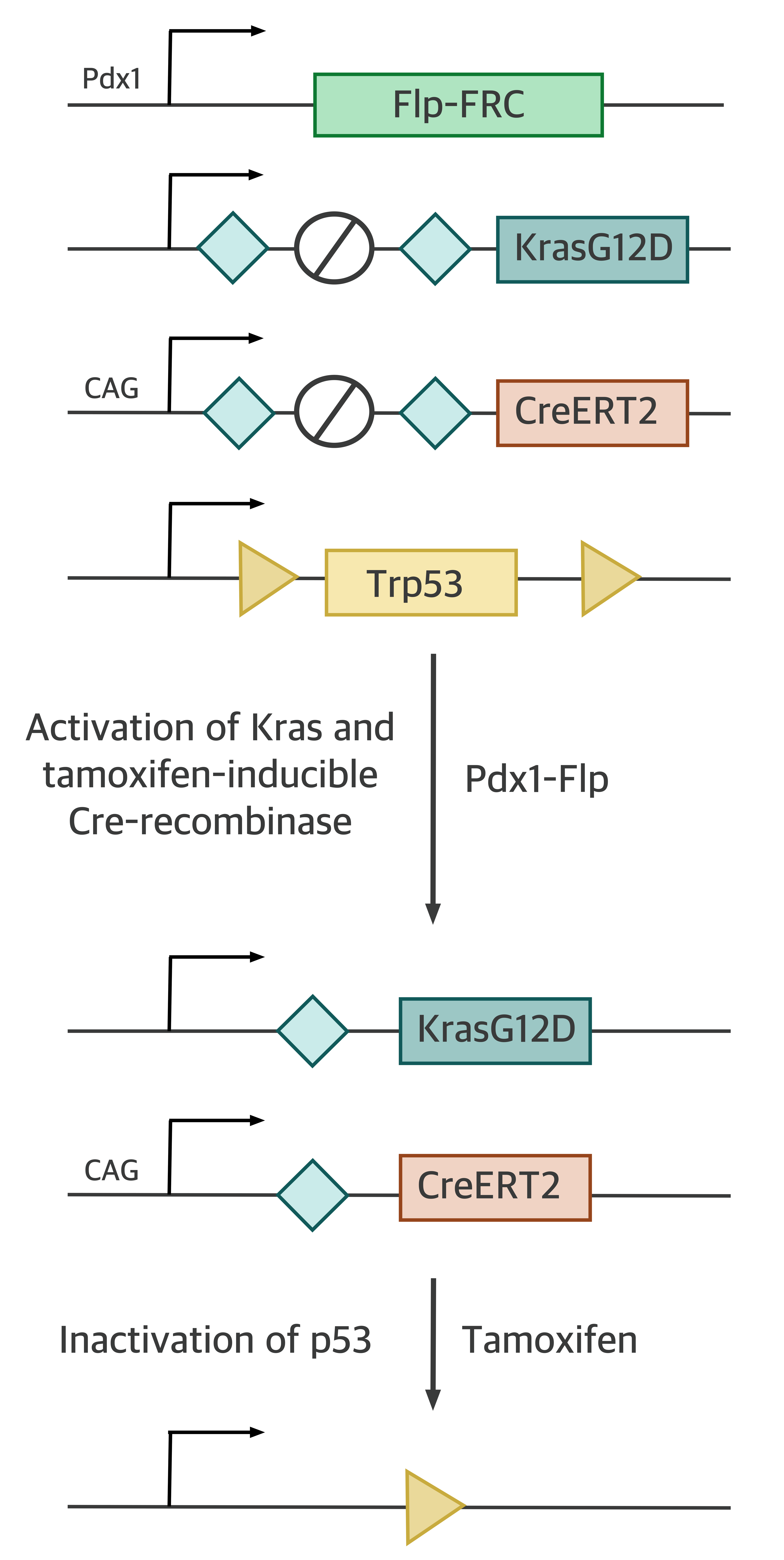
Figure 5. Dual recombination for time-specific p53 inactivation in established KrasG12D-induced PanIN lesions and PDAC cells in the Pdx1-Flp lineage by tamoxifen-mediated activation of CreERT2.
V. In Situ Electroporation: Rapid and Cost-Saving Generation of Transgenic Murine Models
Mouse transgenesis has provided fundamental insights into pancreatic tumorigenesis and has been instrumental for preclinical investigation of novel cancer therapies, but is inherently limited by the long time frames needed to generate and intercross genetically modified mice (91, 92). Furthermore, the multifocal character of PCa in GEMMs strongly contradicts the localized tumor core in the human setting, rendering these models not suitable for surgical interventions (32). To circumvent these limitations, Gürlevik et al. established an in situ electroporation (EP) technique for locally restricted transfection of oncogenic plasmids into the pancreatic tail to induce a single pancreatic tumor nodule (21, 32). The initial KPfl model was obtained via in situ EP of a Cre-expressing plasmid into the pancreas of KRASLSL-G12D/+Trp53Δ/Δmice, leading to focal KRASG12V expression and biallelic loss of Trp53 (32). In a second attempt, mutant KRAS was expressed in p53fl/fl mice using a SB transposase resulting in somatic integration of KRASG12V into the genome and Cre-mediated conditional loss of Trp53 in the electroporated area (Pfl mice) (32). Focal tumor formation at the transfection site was reliably observed using both approaches (32). The KPfl mice presented longer survival (63 to 105 days) compared to the Pfl mice that merely survived between 29 to 37 days and were characterized by poorly differentiated tumors (21). To accelerate metastasis, the investigators additionally co-delivered a transposon encoding a constitutively active form of Akt2 (myrAkt2) together with the Cre-recombinase into the pancreas of KRASLSL-G12D/+Trp53Δ/Δmice, generating KPfl+Akt2 mice (21, 32). Pancreatic tumors that expressed myrAkt2 resembled the human disease and exhibited local infiltration of the surrounding tissue and intratumoral nerves and became widely metastatic (32). Furthermore, to facilitate in vivo imaging, a luciferase-expressing transposon was co-delivered in the Pfl (± Akt2) models, which enabled the accurate monitoring of tumor spreading to distant sites via in vivo imaging spectrometry. These models of resectable transgenic mice develop singular tumor nodules that can be surgically resected to achieve an R0-status and not only represent a unique opportunity to study genetic alterations driving tumor recurrence, but also qualify as a new platform for preclinical screening of novel perioperative, neoadjuvant or adjuvant human-like treatment strategies in PCa (21, 32).
The potential of intrapancreatic plasmid EP was confirmed by a different approach using CRISPR/Cas9-multiplexing transfection for multiplex gene editing in mice. This novel technique allowed high-throughput functional cancer genome analyses, negative-selection screening, and chromosome engineering in murine PCa (67). Using programmable single guide RNAs (sgRNAs), the endonuclease Cas9 can be targeted to a desired genomic locus to locally induce DNA double-strand breaks. These breaks are then imperfectly repaired, which can be exploited to induce indels for gene inactivation (67). This model mediated a dramatic acceleration of tumorigenesis in KC mice with animals starting to succumb to PCa 4 weeks after EP and a tumor incidence of 54% after 24 weeks. The resulting tumors displayed a wide range of histopathologic characteristics from well/moderately differentiated to undifferentiated tumors and presented liver metastases (67). The mosaic pattern of transfection-based CRISPR/Cas9 delivery faithfully reproduces the stochastic nature of human pancreatic tumorigenesis, demonstrating its suitability for phylogenetic research, evading germline genetic engineering and years of interbreeding (67).
VI. In vivo CRISPR-Cas9-Mediated Somatic Recombination
As an alternative mode of rapid, transgenic PDAC generation, Ideno et al. employed adeno-associated virus (AAV) mediated delivery of multiplexed guide RNAs (sgRNAs) to the adult murine pancreas of p48-Cre; LSL-Cas9 mice (45). Here, they demonstrated expression of oncogeni KrasG12D allele through homology-directed repair (HDR), in conjunction with CRISPR-induced excision of cooperating alleles (Trp53, Lkb1 and Arid1A) (45). In analogy with other GEMMs of PDAC, these mice exhibited a spectrum of precursor lesions (pancreatic intraepithelial neoplasia/PanIN, or Intraductal papillary mucinous neoplasm/IPMN) with eventual progression to PDAC (45).
VII. Implantation Models
By the locations of implanted tumor or tumor cells, human xenograft mouse models are two types: heterotopic and orthotopic. In orthotopic xenograft models, tumors or tumor cells are implanted or injected into the equivalent organ from which the cancer originated. The orthotopic xenograft models have similar tumor microenvironment as the original tumor and are deemed to more closely resemble the natural tumorigenesis in human. Subcutaneous xenograft mouse model rarely generates metastases, thus orthotopic mouse models are better suited for such reason. Creating orthotopic pancreatic cancer mouse models is labor-intensive and technically challenging, requires complex imaging to monitor growth of the implanted tumors. Orthotopic implantation of tumor cells or mass into the pancreas. If surgery is necessary to implant tumor cells, the process may require lengthy recovery. Ultrasound-guided injection of tumor cells into the pancreas for the development of orthotopic pancreatic cancer mouse model is less traumatic than surgery (89).
Pancreatic cancer can be induced using in situ injection or pancreatic capsule implantation of tumor cells. A tumor can grow in a month following subcutaneous injection of tumor cells. The tumor is excised and cut into pieces up to 2 mm3. If capsule implantation is used, in the recipient mice the pancreatic capsule is opened, and the tumor implanted into the tail of the gland. The tumor formation period is 4 weeks, and the rate is 100%. Injection of tumor cell suspension has a lower tumor formation rate, the injection port may cause cell shedding, resulting in extensive transplantation metastasis. This method is thus infrequently used. A more contemporary technology for tumor growth involves a thermosensitive biogel. The gel is liquid at a low temperature and turns into jelly at body temperature, which prevents cell shedding. The cells will develop into tumors while lodged on the biogel. In situ tumor formation in pancreatic cancer can fully simulate the internal environment of tumorigenesis and development. The tumorigenesis time and rate are short with in situ growth, and the original tumor structure is maintained, as are most biological characteristics of the human tumor, including growth, local invasion, and distant metastasis. The model is important for studying the tumor microenvironment (55).
Immunodeficient mouse models of orthotopic implantation do not accurately replicate tumorigenesis in humans. Host immune cells in the tumor microenvironment play critical roles in the progression and metastasis of pancreatic tumor. Xenografting human cancer cells directly into an immunocompetent murine host results in graft rejection. Genetically engineered mouse models have been validated. These follow the full spectrum of pancreatic tumorigenesis in humans. Pancreatic tumors from these models can be a good source for the generation of syngeneic orthotopic pancreatic mouse models in an immunocompetent mouse host. Syngeneic orthotopic pancreatic mouse models bring the advantages of orthotopic mouse models and genetically engineered mouse models and are a lower cost alternative to genetically engineered mouse models (89).
The selection of appropriate types of immunodeficient mice (nude mice, SCID, NOD/SCID) for orthotopic pancreatic cancer mouse models depends on the experimental designs. Athymic nude mice (only T-cell deficient) have been widely used for the establishment of orthotopic and heterotopic human pancreatic cancer xenograft mouse models, especially from established human pancreatic cancer cell lines (26). Athymic nude mice are easy to breed and inexpensive. The use of NOD/SCID mice requires less tumor cell inoculums and offers easy tumor formation (90), however, the use of combined immunodeficient mice are limited by the relatively high cost and increased risks of surgical, anesthetic, and infectious complications. The cell viability of tumor cell lines must be assessed by trypan blue exclusion before inoculation. More than 95% of the cells for injection should be viable. Any cell line used for implantation should be routinely tested for mycoplasm contamination to prevent skewed experimental results and animal infection. For human pancreatic cancer cell lines, a single inoculation of 5 × 105 to 1 × 106 cells in serum-free media like PBS is is a good starting number, yet the number of the cells also depends on the cell type and the experimental question. (113). Typically, you need to prepare at least twice the amount of cell suspension for the experiment. A critical step in this operation is to minimize leakage of cancer cells from the injection site, which could result in peritoneal spread. Several other approaches have been reported to reduce this occurrence, including using 30 G fine needle for injection, reducing the injection volume, mixing tumor cell suspensions with 1% Matrigel, and pressing the injection site with a cotton wool tip or with cotton swab immersed with Matrigel for about 1 min after injection (71, 85).
Growth of a tumor can be monitored weekly by ultrasound or by Magnetic resonance imaging (MRI) in orthotopic pancreatic tumor mouse models (85). MRI to monitor pancreatic tumor development, growth, and metastasis for long-term follow-up in preclinical studies, especially with a large number of mice, is extremely expensive and time-consuming (about 1 h for scanning one mouse). If pancreatic cancer cell line is stably transfected with a luciferase-expression construct, the tumor burden including metastasis could be monitored by measuring bioluminescence emission using IVIS (56). Palpable growth of patient tumor in mice may take 4–30 weeks with an average time 14 weeks.
Pancreatic orthotopic implantation steps
- Anesthesia and Analgesia
Use a 2 mL ketamine injection and 0.42 mL xylazine injection (20 mg/mL) mixed in 5.91 sterile injection water or saline at a dose volume of 0.06–0.1 mL/20-25 g body weight. NOTE: According to animal welfare, analgesia is necessary both pre and post operation. 0.05–0.1 mg buprenorphine /kg, SC. The first dose is pre operation and then dosed 3 times every 4 hours post operation continually.
- Surgical operation for orthotopic implantation:
- Anesthetize mice via intramuscular injection (IM) per Step 2.2.1.
- After the animals are fully anesthetized, fix the mice on an experiment board in the right lateral position.
- Keep the mice in the right lateral position. Disinfect the skin around the spleen with iodine then de-iodinate with 75% ethyl alcohol.
- Find the medium point of the spleen and make a 1 cm vertical incision on abdomen to expose the spleen.
- Draw out a part of pancreas tissue under the spleen gently with flat-tip tweezers, and suture a mouse homograft tumor piece from seed mouse on the pancreas of recipient mouse by 9-0 Absorbable surgical suture.
- Close the abdomen with a 6-0 silk suture by double seam. Achieve homeostasis by compression.
- After finishing tumor implantation, if neither bleeding nor tumor tissue leakage occurs, keep the animals in a warm cage.
- Monitor the animal until it regains sufficient consciousness to maintain sternal recumbency; return the animal to the animal room after full recovery from the anesthesia. Monitor the tumor bearing mice by palpating the abdomen near the spleen and select out the mice bearing orthotopic tumors (3)
One alternative to the spontaneous KPC mouse model is to use an orthotopic implantation model of PDA (22). The direct surgical implantation of tumor cell lines in to the native tissue site is a more cost-effective and predictable method of recapitulating the tissue-specific tumor microenvironment (TME) of PDA. Pancreatic tumor implantation requires a labor-intensive surgical procedure that introduces aberrant inflammation at the suture site in the abdominal wall, and includes a lengthy post-operative recovery(89, 107).
Ultrasound-guided imaging allows visualization of the injection needle in the peritoneal cavity, while implanting tumor cells into the pancreas, thus avoiding surgical implantation and associated complications. This approach, termed ultrasound-guided orthotopic tumor implantation model (UG-OTIM) has been previously established in a xenograft models of pancreatic cancer(44). To determine if there was a benefit to using the UG-OTIM approach rather than the traditional surgical orthotopic model, a comparison of the seeding of PDA tumors in the peritoneal wall of mice was evaluated after each procedure. The rate of seeding additional tumors in the peritoneum is greatly reduced in the UG-OTIM method as compared to surgical implantation. The use of high-resolution ultrasonography to direct implantation of murine PDA cell lines to the autochthonous tissue site is a reliable alternative to both the KPC and surgical orthotopic models (34).
A comparative study looked at a pancreatic adenocarcinoma model based on tumor injection into the pancreatic head v tail models in C57/BL6 mice. Pancreatic head and tail orthotopic cancer models produce consistent tumors, but the patterns of tumor spread and survival differ according to the site of injection. The overall survival of animals at 40 days following tumor induction was significantly lower in the pancreatic head injection group. Multiple liver metastases were noted 50% animals in the head injection group, without evidence of peritoneal metastases. In the pancreatic tail injection group, 90% animals had multiple peritoneal metastases, 45% animals had evidence of isolated liver deposits. Tumors in both regions of the pancreas had similar histologic characteristics, with a dense fibrotic stroma at the interface between the tumor and the normal pancreas (79).
In a study by Lei Dai, et al. two orthotopic xenograft models were developed, in which either a tumor mass or Matrigel-tumor cell mixture was directly implanted into the pancreas of mice (18). The findings showed that the orthotopic tumor mass implantation model had superior performance results than the other models in terms of tumor volume and metastasis. Using an in vivo imaging system, the local invasion and metastasis of the tumors were observed in the orthotopic tumor mass implantation and Matrigel block implantation models, but not in the subcutaneous xenograft mice. At day 36, the tumor-bearing mice were sacrificed and examined for metastatic tumors. In the orthotopic tumor mass xenograft model, 80% of the mice exhibited tumor metastasis, with the majority exhibiting peritoneal metastasis. Similarly, in the orthotopic Matrigel block xenograft model, 80% of the mice exhibited tumor metastasis, but the metastatic sites were slightly different, with a higher prevalence of peritoneal and lower frequency of liver metastasis. No tumor metastasis was identified in the subcutaneous xenograft model. Tumor cells were harvested using 0.05% trypsin solution, washed in phosphate buffered saline (PBS) (Sigma Aldrich), and re-suspended as a single-cell suspension in DMEM. The cell viability was greater than 95% when tested using the trypan blue exclusion. A concentration of 5 × 106 cell/ml PAN02 cells was mixed with ice-cold MatrigelTM matrix solution (BD Biosciences, North Ryde, Australia) in a ratio of 1:1. Each animal was injected with 2.5 × 105 viable cells in 50 μl of MatrigelTM (18).
Utilizing subclones with pure epithelial morphology (referred to as LM-P), William Tseng et al. developed and characterized an immunocompetent, orthotopic mouse model of pancreatic cancer in which disease develops consistently and with rapid and predictable growth kinetics. Eight to ten-week-old immunocompetent female B6/129 mice, histocompatible (H-2) with the tumor cells used in this study, were obtained from Jackson Laboratories (Bar Harbor, ME). After either injection of suspended LM-P cells into the pancreas, or implantation of a subcutaneous LM-P tumor fragment onto the pancreas, tumors were noted as early as two weeks and progressed predictably by four weeks. Comparison of average tumor volumes at two and four weeks demonstrated no statistically significant differences, suggesting similar growth kinetics with either technique. By six weeks, tumors in some mice became difficult to measure owing to their extensive, locally invasive nature and poorly defined borders. At this stage in disease progression, liver metastases were also frequently noted and mice appeared ill. Of note, some mice also developed biliary and gastric outlet obstruction during the course of disease progression. By eight weeks, mortality was consistently 100%.
Pancreatic tumors developed in 100% of mice following orthotopic implantation of LM-P cells. Of these mice, 90% developed liver metastases while no mice developed peritoneal carcinomatosis or hemorrhagic ascites. No significant differences were noted in the frequencies of uptake and metastases with either technique. Lung metastases were noted in some mice, particularly those with more advanced disease; however, the frequency of metastasis to this organ was not systematically evaluated. Pancreatic tumors and liver metastases also developed with fewer than 106 cells injected, but tumor volumes were initially smaller and disease progression occurred more slowly; in contrast, the use of cells passaged more frequently in vitro generated pancreatic tumors with more rapid disease progression. Furthermore, the disease develops rapidly and predictably. This orthotopic mouse model of pancreatic cancer is established by surgical implantation of tumor cells into the pancreas of an immunocompetent host (114).
Boj et al. established organoid models from normal and neoplastic murine and human pancreas tissues. Pancreatic organoids can be rapidly generated from resected tumors and biopsies, survive cryopreservation and exhibit ductal- and disease stage-specific characteristics. Orthotopically transplanted neoplastic organoids recapitulate the full spectrum of tumor development by forming early-grade neoplasms that progress to locally invasive and metastatic carcinomas. Due to their ability to be genetically manipulated, organoids are a platform to probe genetic cooperation. Comprehensive transcriptional and proteomic analyses of murine pancreatic organoids revealed genes and pathways altered during disease progression. The confirmation of many of these protein changes in human tissues demonstrates that organoids are an useful model to study pancreatic adenocarcinoma. Pancreatic organoids derived from wild-type mice and PDA GEMMs accurately recapitulate physiologically relevant aspects of disease progression in vitro. Following orthotopic transplantation, organoids from wild-type mouse normal pancreata are capable of regenerating normal ductal architecture, unlike other 3D model systems. We further developed methods to generate pancreatic organoids from normal and diseased human tissues, as well as from endoscopic needle biopsies. Following transplantation, organoids derived from murine and human PDA generate lesions reminiscent of PanIN and progress to invasive PDA. Organoids are useful to identify molecular pathways that correlate with disease progression, and that represent therapeutic and diagnostic opportunities (8).
VIII. Patient-Derived Xenografts of Pancreatic Cancer
Patient-derived xenograft (PDX) mouse models certainly play a significant role in evaluation of chemotherapies, with the human aspect of these mice being the human tumor. Yet these PDX mice contribute in other profound ways including improved understanding of metabolism and a platform for improved genomic/transcriptomic analyses. Within this framework and in the context of PDAC, drug delivery, etiology, epigenetic phenomena, detection, prognosis and some technical aspects will be considered. Thus, the content of this subsection will focus on these concepts and stay within the bounds of themes geared toward translational impact.
A. Therapy
Models that harbor cancers developed from human sources are ideal platforms for evaluating novel therapeutic regimens and can recapitulate responses that mirror those observed in the same donor patient, which is a more common feature of all cancer PDX models (49). PDX models represent an in vivo system developed for a more personalized therapy consistent with precision medicine initiatives. Generation of multiple PDX models for a single study would be a true preclinical assessment that best mimics a clinical trial due to the variety of individual PDAC tumors in these mice. In this manner, multiple therapeutic regimens have been assessed in PDX PDAC mice as demonstrated in Table 4, though more recently a litany of novel approaches have been performed including modified drug derivatives, natural products, viral therapies, radiation, chemoresistance blockers, and metabolic inhibitors as shown in the following examples.
Table 4. Recent and more novel therapeutic assessments in PDAC PDX mice.

The addition of a stearate to gemcitabine (GEM) generated a fatty acid GEM derivative termed stearoyl gemcitabine (4NSG) that demonstrated more potent anticancer effects: greatly reduced cell proliferation, enhanced apoptosis and induced cytotoxicity in 2D and 3D PDAC cell cultures. This was recapitulated in PDX models of PDAC which also exhibited reduced angiogenic potential via dramatically reduced VEGF and a concomitant reduction in tumor volume compared to untreated and GEM-HCl-treated mice (47). A second approach utilized a natural product, Brucia javanica (also Brucea), a medicinal plant with anti-inflammatory and anti-oxidant properties, in combination with GEM to improve its efficacy in vivo as assessed in orthotopically-implanted PDX mice. These mice had reduced PC tumor growth, increased apoptosis and an overall improvement in survival (130). Building on a relatively new standard of care using GEM plus nab-paclitaxel (GEM+NP), a NOTCH-sensitive oncolytic adenovirus (AduPARE1A) was introduced into PDAC PDX mice that enhanced the anti-cancer effect of GEM+NP. This was evident in a significant reduction in the number of PC stem cells (PCSCs), number of tumorspheres grown in an anchorage-independent manner, and tumor volume while maintaining a low toxic profile (68). This same adenovirus will be highlighted in PC patient-derived organoid (PDO) mice in the next section.
Other therapeutic regimens aimed to introduce more novel and potentially dual threat approaches or improve existing drug efficacy by blocking mechanisms of chemoresistance. For the former, the beta-emitting 177Lu-4A06 radioisotope, an antibody that recognizes the CUB domain containing protein 1 (CDCP1) was dosed into PC PDX mice and generated significantly reduced tumor growth and volume leading to improved survival. Not only was this a product of cytotoxic destruction of CDCP1 positive cells but likely included improved radioimmunotherapy due to less influence of and improved delivery through the dense tumor stroma of PC (74). Additional studies in PDAC PDX mice demonstrated prevention of chemoresistance from DNA-damaging agents (like GEM) and microtubule poisons (like NP) via CDK4/6 inhibition (99). Finally, using three unique PC PDX mouse lines, a unique PGAM1 inhibitor (KH3) was evaluated in these mice and proven to downregulate glycolysis, mitochondrial respiration and glutathione metabolism leading to cell cycle arrest, increased apoptosis and reduced tumor volume (125).
Despite advancing science with these PDX models in regards to therapy, the challenge remains whether these mice can be effectively employed to improve outcomes for the patient for whose tumor the mice harbor. None of these reports demonstrated direct impact of the approach tested in mice in the patient who provided the primary tumor. Indeed, this is a formidable task since development of PDAC PDX mice in a multiplicity of animals with the same tumor requires at least a few rounds of passaging to characterize tumor kinetics and survival prior to assignment to a treatment arm. Other limitations exist including the use of immunocompromised mice to house these tumors, and though patients often present with compromised immune function to the advantage of cancer spread, the level of whole body immunosuppression is likely greater in the mouse. Indeed, mouse models work best in tandem with other models, and for PDAC PDX mice, this would include autochthonous GEM mice and orthotopic PC implantation in syngeneic mice as previously discussed.
B. Metabolism
Since many studies with PDAC PDX mice are focused on therapeutic interventions, primarily through small molecule inhibitors, it is important to consider points related to drug metabolism and indeed other metabolic considerations that could impact drug response and/or tumor development. This is particularly relevant in these PDX models since they represent a more human cancer as it expands in an in vivo platform. So the first consideration will be focused on drug metabolism followed by examples of other metabolic pathways including those pertaining to carbohydrates, lipids, amino acids and nucleic acids.
Drug metabolism: PDAC PDX mice present a great system for improved understanding of drug behavior including small molecule uptake and target engagement using titrated concentrations of a novel fluorescently-labeled GEM conjugate. Such an approach generated a dose response in fluorescence intensity that was significantly higher than PDX mice administered the fluorophore alone (no GEM), though highest levels of drug uptake correlated with areas of necrosis as revealed by tissue morphology. And this methodology can also be applied to other drugs for similar evaluations (106). [18F]-FAC metabolic PET imaging was as effective in measuring GEM uptake as [14C]-GEM in 3 individual PDAC PDX mouse models, demonstrating [18F]-FAC metabolic PET as a solid surrogate of GEM uptake (98). However, the above fluorophore assessment fell short of providing real evidence of metabolic changes related to drug kinetics. Studies that focus on pharmacokinetics/pharmacodynamics (PK/PD) provide more relevant information regarding plasma concentration of active drug. In examination of dexamethasone (DEX) in PDAC PDX mice, the greatest linear inhibitory function of DEX was highest in PDX mice compared to those established with PC cell lines and at times, twice as high depending on the PC cell line (131). Employing a similar PK/PD evaluation to assess a novel liposome formulation loaded with doxorubicin (Dox) and a photosensitizer, irradiation in PDAC PDX mice generated a 12-fold higher influx of drug into tumor than irradiated PDX mice without liposome administration. This amounted to a 7-fold higher Dox concentration in the tumor over time with a 4-fold reduction in tumor volume(61). These studies are similar to older published works that describe features of PK/PD for other drugs as exemplified with the Hedgehog inhibitor TAK-411 (53). But there remains a paucity for the examination of drug metabolites in vivo like that recently completed for gemcitabine in KC and KPC GEM mice (as previously described) (9) and done two decades ago in preclinical models for drugs like cyclophosphamide and ifofamide by addition of a CP450 gene-directed enzyme prodrug therapy (P450 GDEPT) (12). So to a great extent, following drug metabolites will best reflect drug efficacy and toxicity as a support for current and future clinical trials.
Energy Sources: Beyond the impact of drug metabolism is how other pathways are altered during the etiology and treatment of PC, which would include those focused on tumor energy sources like sugar/carbohydrates and lipids as well as modifications that alter resources for gene expression at both the transcriptional (nucleic acids) and translational (amino acids) levels. A rigorous study focused on the Warburg effect utilized 15 PDAC PDX models (each uniquely generated from 1 of 15 PDAC patients) treated with FX11, a lactate dehydrogenase A inhibitor (LDH-A). These cohorts of PDX mice demonstrated increased apoptosis, reduced proliferation and decreased tumor growth that was restricted to tumors harboring a mutant p53 allele, as FX11 was ineffective in human PC with wild type p53. This work was well represented by 13C MRS imaging modalities to further support these observed effects (94). Another study demonstrated the ability of mIR-7 to inhibit PDAC development in PDX mice by interfering with the Warburg effect via suppression of glycolysis and associated reduction in autophagy (28). Indeed, a high glucose environment has been linked to enhanced tumor growth via sterol regulatory element-binding protein 1 (SREBP1)-induced autophagy (134). And SREBP1 is a potent regulator of lipogenic genes including acetyl-CoA carboxylase (ACC), fatty acid synthase (FASN), and stearoyl-CoA desaturase-1 (SCD1) (108). Since lipids can also serve as an energy source for primary and metastatic PC, it is critical to consider these alternative pathways in PDAC PDX mouse models. This has not been demonstrated to this point in vivo, though de novo lipid synthesis has been shown to contribute to GEM resistance in an orthotopic xenograft mouse model using Panc1 cells (109) and targeting lipid metabolism has abrogated PC development in subcutaneous tumors from MiaPaCa2 cells (78). Regarding amino acid metabolism, there is a clear focus on amino acid transporters across the cell membrane include the SLC family, of which SLC6A14 had the broadest profile for amino acid substrate transport. Using 10 unique PDAC PDX mice, it was established that mRNA levels of SLC6A14 were at least 100-fold higher in 8 of the 10 PDX mouse models (with the other two at 5- and 10-fold). The highest level reached over a 40,000-fold increase, though the SLC6A14 inhibitor alpha-methyltryptophan used to demonstrate anti-cancer effects in PC cell line-derived xenograft mice was not further assessed in these PDAC PDX mice (15). Finally, nucleotide metabolism should not be ignored, as the small molecular inhibitor decitabine impaired pyrimidine biosynthesis and indirectly impact oncogenic KRAS-driven events. This demonstrated the utility of repurposing decitabine as a potential effective means of blocking mutant KRAS addiction in PC (77).
C. Genomics/Transcriptomics
In consideration of the role of nuclei and amino acids is how PDAC PDX mice can provide insight regarding gene expression and the profile to establish cancer subtypes, which is made possible by elegant and rigorous genomics/transcriptomics evaluations. In a project employing 12 unique PDAC PDX models, a predictive and prognostic model of PDAC was discovered following germline BRCA1/2 mutation and its association with homologous recombination deficiency (HRD). This was done by identification of tumor polyploidy and low proliferative index (Ki67), which serve as predictors of low efficacy following treatment with cisplatin and talaxoparib. Tumor polyploidy and basal-like transcriptomic subtype were independent predictors in this 7-arm preclinical trial of over 470 PDX mice (123). Another well-designed study exploited 10 unique PC patient-derived PDX mice for microarray gene expression evaluation where 15 genes had a 5-fold or greater change in expression when compared with normal adjacent tissue. Interestingly, the gene with the highest (23-fold) increase in expression was SLC6A14, the broad amino acid transporter discussed above. Gene ontology analysis from this data showed that genes with at least a 2-fold increase were associated with the cell cycle and mitosis, where over 20 such genes were cell cycle regulators (95). Indeed, this is expected considering the general nature of cancer cells including those from PC.
Beyond increased proliferation, PDAC PDX mice were administered a Msi (Musashi, a RNA binding protein) inhibitor which blocked tumor growth via suppression of stem cells, representing a novel means of preventing chemoresistance in vivo. These findings were further supported by GEM mice with a specific Msi reporter system that allowed monitoring of Msi-positive cells, among a variety of other outstanding modalities (23). This is a departure from standard targets for inhibition as a means of abrogating tumor development. Another unique point of investigation with PDAC PDX mice addressed an epigenetic phenomena demonstrating nicotine induction of IL8 secretion from the stromal compartment and subsequent increased IL8R in PC parenchyma with worsened cancer cachexia and increased tumor mass (116). An additional study focused on cachexia using patient-derived PDAC mice employed genome-wide microarray analysis which demonstrated increased ECM protein-encoding genes in diaphragm that were down-regulated in tibialis anterior muscle, among other notable differences (83). Furthermore, PDAC PDX mice can serve as predictors of disease-free survival (DFS) in surgically resected PDAC patients, where survival was half that (6 vs. 12 months) for patients whose tumor were successfully engrafted compared to those whose tumors failed to take. Also, the pathology and genetics of primary tumors were recapitulated in the PDX-derived tumors (13)
D. PDX Technical Advancements
PDAC PDX mouse models have been improved and/or expanded in a variety of ways. Cryopreserved PDAC was introduced into liver to generate a robust model of PDAC liver metastasis of tumors from 6 of 10 patient donors in one third of recipient mice (110). Another means of advancing PDX models was deriving cancer from a tissue source other than primary tumor. In this regard, patient ascites were successfully used to derive PDAC PDX mice, which interestingly became GEM chemoresistant over time that recapitulated the same response in these patients with PDAC (64). Yet another technical innovation was clear demonstration that the PDAC tumor formation rate of primary PDX (F1) mice was half that of F2 and F3 generated PDX mice, which also had a significantly reduced latency. Here, it is likely that serial transplantation selects for tumor cell clones with higher viability, stemness features, and thus lower similarity to the original tumor. Despite these kinetic changes, all three generations of PDX mice had similar histopathology and increased Ki67 positive proliferative indices to that observed in the primary tumor (128). These technical enhancements in PDAC PDX modelling technology provide the means to: (1) cryopreserve and yet employ PDX lineages, (2) expand from invasive resection of primary tumor to less invasive collection of ascites as a source for deriving PDX mice, and (3) consider use of F2 generations and beyond for development of more efficient PDX systems.
With the advent and progressive utilization of PDAC PDX mice, therapeutic evaluations, improved understanding of cancer metabolism, and identification of genomic/transcriptomic profiles related to PDAC subtypes will greatly assist in exposing novel targets and new therapies to better fight pancreatic cancer. To further assist in this goal, establishing tumors directly from human 3D spheroids generated from isolated PDAC (called patient-derived organoids or PDOs) introduces yet another useful modelling system.
E. PDOX Models of PDAC
There are some distinct advantages to developing patient-derived organoids from PDAC and implanting/injecting them into immunocompromised mice similar to PDX models. This includes having: (1) the option to genetically modify these structures to study additional genetic alterations, (2) a more pure, clonal population of PDAC (less heterogeneity than primary tumor (96)), and (3) a platform to investigate signals and molecular events between human parenchyma and mouse mesenchyma. As a starting point, PDAC organoids recapitulatepatient-specific molecular and histopathologic signatures, making them ideal for improved understanding of the etiology of the patient's tumor, the varied response to therapies, and the utility in predicting drug responses (96).
Introduction of PDOs into mouse flanks or pancreas have been generated but with fairly limited application. Yet, development of xenograft PDAC tumors in PDOX mice have been generated to maintain the histoarchitecture of cancers when compared to primary tumors from corresponding patients (43). The few studies that have employed PDOX mice have not only further supported this model as a useful platform for evaluation but extended these studies in unique ways which correlated to the strengths of this patient-derived in vivo system. One excellent example of this is the evaluation of the impact of oncolytic adenovirus on mice harboring orthotopically implanted PDOs derived from primary tumor and metastases. In this scenario, PDOX mice were preferred over other models including PDX mice since these organoids more readily accepted viral entry of DNA material to demonstrate the viral preference of parenchymal cells compared to other cell types (93). Another unique treatment included a RNA-based therapeutic, an anti-mIR-21 RNA that self assembles into a nanoparticle, called TPN-21, which was delivered twice weekly in PDAC PDOX mice. The end result was that TPN-21 strongly and progressively slowed tumor growth, resulting in a greater than 50% suppression of tumor growth (25). Beyond these few studies, there is a real paucity in the use of PDAC PDOX models for evaluations as described for PDAC PDX mice. However, therapy assessment in PDOX mice from other GI cancers has been employed and should be considered in PDAC PDOX studies. Also, mouse-derived PDOs and their introduction into syngeneic mice have been well-documented in ways that to some regard match that done in PDAC PDX mice. Several of these would be ideal for evaluation in PDAC PDOX mice.
To demonstrate the impact of therapy strictly at the level of developing carcinoma, CRC (colorectal cancer) PDOX mice demonstrated a similar response to therapy as the patients whose tumor was implanted into mice, and this included an initial response to paclitaxel before developing resistance. A similar pattern was observed in regorafenib-sensitive or resistant patients, where the implanted PDO was sensitive or resistant as its patient donor (117). In mouse-derived organoids introduced into obese mice, it was determined that obesity-induced inflammation stimulated tumor progression and metastasis (63). In elegant work done with KPC-derived organoids marked with Tomato Red including organoid co-cultures, metabolic flux was measured via pyruvate carboxylase and malic enzyme 1 activity in parenchymal (cancer cells) and mesenchymal (fibroblasts) cell compartments. The broad result among a myriad of other nuanced findings was that expression of both pyruvate carboxylase and malic enzyme was essential for cancer growth, with details regarding how various cell compartments play a role in this equation (57). With these as druggable targets, organoids have been front and center as a platform for assessing optical imaging of drug-induced metabolic flux (120). Indeed, such approaches should be considered in PDAC PDOX mice.
In order to best recapitulate human PDAC, it is important to consider the use of in vivo models that employ human cancer cells in combination with autochthonous GEM and orthotopic syngeneic models. The combination thereof covers a broad spectrum of systems to best understand the etiology of pancreatic cancer and its response to new and repurposed therapies. Patient-derived PDAC as primary or passaged cancer cells (PDX mice) or those allowed to spontaneously develop into 3D structures (PDOX mice) can be introduced into immunocompromised mice and then employed in a variety of studies to evaluate therapies, assess metabolic changes, and determine genomic/transcriptomic profiles. This has been done to some extent in PDAC PDX mice with yet only very modest considerations utilizing PDAC PDOX mice. In part, this section was written to encourage additional investigations in PDAC PDOX mice, as such studies will advance our understanding of how best to attack pancreatic cancer in humans.
IX. References
- Adrian K, Strouch MJ, Zeng Q, Barron MR, Cheon EC, Honasoge A, Xu Y, Phukan S, Sadim M, Bentrem DJ, Pasche B, and Grippo PJ. Tgfbr1 haploinsufficiency inhibits the development of murine mutant Kras-induced pancreatic precancer. Cancer Res 69: 9169-9174, 2009. PMID: 19951995.
- Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, and DePinho RA. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev 17: 3112-3126, 2003. PMID: 14681207.
- An X, Ouyang X, Zhang H, Li T, Huang Y-Y, Li Z, Zhou D, and Li Q-X. Immunophenotyping of Orthotopic Homograft (Syngeneic) of Murine Primary KPC Pancreatic Ductal Adenocarcinoma by Flow Cytometry. J Vis Exp 140:57460,2018. PMID: 30371656.
- Awasthi N, Kronenberger D, Stefaniak A, Hassan MS, von Holzen U, Schwarz MA, and Schwarz RE. Dual inhibition of the PI3K and MAPK pathways enhances nab-paclitaxel/gemcitabine chemotherapy response in preclinical models of pancreatic cancer. Cancer Lett 459: 41-49, 2019. PMID: 31153980.
- Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U, Hanahan D, Redston MS, Chin L, and Depinho RA. Both p16Ink4a and the p19Arf-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci U S A 103: 5947-5952, 2006. PMID: 16585505.
- Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P, Hezel AF, Horner J, Lauwers GY, Hanahan D, and DePinho RA. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev 20: 3130-3146, 2006. PMID: 17114584.
- Bardeesy N, Morgan J, Sinha M, Signoretti S, Srivastava S, Loda M, Merlino G, and DePinho RA. Obligate roles for p16Ink4a and p19Arf-p53 in the suppression of murine pancreatic neoplasia. Mol Cell Biol 22: 635-643, 2002. PMID: 11756558.
- Boj SF, Hwang CI, Baker LA, Chio, II, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, Gracanin A, Oni T, Yu KH, van Boxtel R, Huch M, Rivera KD, Wilson JP, Feigin ME, Ohlund D, Handly-Santana A, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 160: 324-338, 2015. PMID: 25557080.
- Buchholz SM, Goetze RG, Singh SK, Ammer-Herrmenau C, Richards FM, Jodrell DI, Buchholz M, Michl P, Ellenrieder V, Hessmann E, and Neesse A. Depletion of Macrophages Improves Therapeutic Response to Gemcitabine in Murine Pancreas Cancer. Cancers (Basel) 12: 2020. PMID: 32698524.
- Carriere C, Gore AJ, Norris AM, Gunn JR, Young AL, Longnecker DS, and Korc M. Deletion of Rb accelerates pancreatic carcinogenesis by oncogenic Kras and impairs senescence in premalignant lesions. Gastroenterology 141: 1091-1101, 2011. PMID: 21699781.
- Cash TP, Alcala S, Rico-Ferreira MDR, Hernandez-Encinas E, Garcia J, Albarran MI, Valle S, Munoz J, Martinez-Gonzalez S, Blanco-Aparicio C, Pastor J, Serrano M, and Sainz B, Jr. Induction of Lysosome Membrane Permeabilization as a Therapeutic Strategy to Target Pancreatic Cancer Stem Cells. Cancers (Basel) 12: 2020. PMID: 32635473.
- Chen L, and Waxman DJ. Cytochrome P450 gene-directed enzyme prodrug therapy (GDEPT) for cancer. Curr Pharm Des 8: 1405-1416, 2002. PMID: 12052216.
- Chen Q, Wei T, Wang J, Zhang Q, Li J, Zhang J, Ni L, Wang Y, Bai X, and Liang T. Patient-derived xenograft model engraftment predicts poor prognosis after surgery in patients with pancreatic cancer. Pancreatology 20: 485-492, 2020. PMID: 32113935.
- Choi EA, Choi Y-S, Lee EJ, Singh SR, Kim SC, and Chang S. A pharmacogenomic analysis using L1000CDS2 identifies BX-795 as a potential anticancer drug for primary pancreatic ductal adenocarcinoma cells. Cancer Lett 465: 82-93, 2019. PMID: 31404615.
- Coothankandaswamy V, Cao S, Xu Y, Prasad PD, Singh PK, Reynolds CP, Yang S, Ogura J, Ganapathy V, and Bhutia YD. Amino acid transporter SLC6A14 is a novel and effective drug target for pancreatic cancer. Br J Pharmacol 173: 3292-3306, 2016. PMID: 27747870.
- Copeland NG, and Jenkins NA. Harnessing transposons for cancer gene discovery. Nat Rev Cancer 10: 696-706, 2010. PMID: 20844553.
- Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH, Levy DE, Settleman J, Engelman JA, and Bardeesy N. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res 71: 5020-5029, 2011. PMID: 21586612.
- Dai L, Lu C, Yu XI, Dai LJ, and Zhou JX. Construction of orthotopic xenograft mouse models for human pancreatic cancer. Exp Ther Med 10: 1033-1038, 2015. PMID: 26622435.
- David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, Iacobuzio-Donahue CA, and Massague J. TGF-β Tumor Suppression through a Lethal EMT. Cell 164: 1015-1030, 2016. PMID: 26898331.
- DeCant BT, Principe DR, Guerra C, Pasca di Magliano M, and Grippo PJ. Utilizing past and present mouse systems to engineer more relevant pancreatic cancer models. Front Physiol 5: 464, 2014. PMID: 25538623.
- Demir IE. Transcriptional and functional characterization of the first neuro-invasive, genetically engineered mouse model of pancreatic cancer. 2015.
- Foster DS, Jones RE, Ransom RC, Longaker MT, and Norton JA. The evolving relationship of wound healing and tumor stroma. JCI Insight 3: 2018. PMID: 30232274.
- Fox RG, Lytle NK, Jaquish DV, Park FD, Ito T, Bajaj J, Koechlein CS, Zimdahl B, Yano M, Kopp J, Kritzik M, Sicklick J, Sander M, Grandgenett PM, Hollingsworth MA, Shibata S, Pizzo D, Valasek M, Sasik R, Scadeng M, et al. Image-based detection and targeting of therapy resistance in pancreatic adenocarcinoma. Nature 534: 407-411, 2016. PMID: 27281208.
- Gao M, Lin M, Moffitt RA, Salazar MA, Park J, Vacirca J, Huang C, Shroyer KR, Choi M, Georgakis GV, Sasson AR, Talamini MA, and Kim J. Direct therapeutic targeting of immune checkpoint PD-1 in pancreatic cancer. Br J Cancer 120: 88-96, 2019. PMID: 30377341.
- Gilles ME, Hao L, Huang L, Rupaimoole R, Lopez-Casas PP, Pulver E, Jeong JC, Muthuswamy SK, Hidalgo M, Bhatia SN, and Slack FJ. Personalized RNA Medicine for Pancreatic Cancer. Clin Cancer Res 24: 1734-1747, 2018. PMID: 29330203.
- Gray MJ, Wey JS, Belcheva A, McCarty MF, Trevino JG, Evans DB, Ellis LM, and Gallick GE. Neuropilin-1 suppresses tumorigenic properties in a human pancreatic adenocarcinoma cell line lacking neuropilin-1 coreceptors. Cancer Res 65: 3664-3670, 2005. PMID: 15867361.
- Grippo PJ, Nowlin PS, Demeure MJ, Longnecker DS, and Sandgren EP. Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant Kras in transgenic mice. Cancer Res 63: 2016-2019, 2003. PMID: 12727811.
- Gu DN, Jiang MJ, Mei Z, Dai JJ, Dai CY, Fang C, Huang Q, and Tian L. microRNA-7 impairs autophagy-derived pools of glucose to suppress pancreatic cancer progression. Cancer Lett 400: 69-78, 2017. PMID: 28450156.
- Guerra C, and Barbacid M. Genetically engineered mouse models of pancreatic adenocarcinoma. Mol Oncol 7: 232-247, 2013. PMID: 23506980.
- Guerra C, Collado M, Navas C, Schuhmacher AJ, Hernandez-Porras I, Canamero M, Rodriguez-Justo M, Serrano M, and Barbacid M. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 19: 728-739, 2011. PMID: 21665147.
- Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, Dubus P, Sandgren EP, and Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 11: 291-302, 2007. PMID: 17349585.
- Gurlevik E, Fleischmann-Mundt B, Brooks J, Demir IE, Steiger K, Ribback S, Yevsa T, Woller N, Kloos A, Ostroumov D, Armbrecht N, Manns MP, Dombrowski F, Saborowski M, Kleine M, Wirth TC, Oettle H, Ceyhan GO, Esposito I, Calvisi DF, et al. Administration of Gemcitabine After Pancreatic Tumor Resection in Mice Induces an Antitumor Immune Response Mediated by Natural Killer Cells. Gastroenterology 151: 338-350 e337, 2016. PMID: 27210037.
- Habbe N, Shi G, Meguid RA, Fendrich V, Esni F, Chen H, Feldmann G, Stoffers DA, Konieczny SF, Leach SD, and Maitra A. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A 105: 18913-18918, 2008. PMID: 19028870.
- Hay CA, Sor R, Flowers AJ, Clendenin C, and Byrne KT. Ultrasound-Guided Orthotopic Implantation of Murine Pancreatic Ductal Adenocarcinoma. J Vis Exp 2019. PMID: 31814615.
- He M, Henderson M, Muth S, Murphy A, and Zheng L. Preclinical mouse models for immunotherapeutic and non-immunotherapeutic drug development for pancreatic ductal adenocarcinoma. Ann Pancreat Cancer 3: 2020. PMID: 32832900.
- Heid I, Lubeseder-Martellato C, Sipos B, Mazur PK, Lesina M, Schmid RM, and Siveke JT. Early requirement of Rac1 in a mouse model of pancreatic cancer. Gastroenterology 141: 719-730, 730 e711-717, 2011. PMID: 21684285.
- Heiser PW, Cano DA, Landsman L, Kim GE, Kench JG, Klimstra DS, Taketo MM, Biankin AV, and Hebrok M. Stabilization of beta-catenin induces pancreas tumor formation. Gastroenterology 135: 1288-1300, 2008. PMID: 18725219.
- Hezel AF, Gurumurthy S, Granot Z, Swisa A, Chu GC, Bailey G, Dor Y, Bardeesy N, and Depinho RA. Pancreatic LKB1 deletion leads to acinar polarity defects and cystic neoplasms. Mol Cell Biol 28: 2414-2425, 2008. PMID: 18227155.
- Hill R, Calvopina JH, Kim C, Wang Y, Dawson DW, Donahue TR, Dry S, and Wu H. PTEN loss accelerates KrasG12D-induced pancreatic cancer development. Cancer Res 70: 7114-7124, 2010. PMID: 20807812.
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, Wright CV, Hruban RH, Lowy AM, and Tuveson DA. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4: 437-450, 2003. PMID: 14706336.
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, and Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7: 469-483, 2005. PMID: 15894267.
- Hiroshima Y, Maawy AA, Katz MH, Fleming JB, Bouvet M, Endo I, and Hoffman RM. Selective efficacy of zoledronic acid on metastasis in a patient-derived orthotopic xenograph (PDOX) nude-mouse model of human pancreatic cancer. J Surg Oncol 111: 311-315, 2015. PMID: 25394368.
- Huang L, Holtzinger A, Jagan I, BeGora M, Lohse I, Ngai N, Nostro C, Wang R, Muthuswamy LB, Crawford HC, Arrowsmith C, Kalloger SE, Renouf DJ, Connor AA, Cleary S, Schaeffer DF, Roehrl M, Tsao MS, Gallinger S, Keller G, et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat Med 21: 1364-1371, 2015. PMID: 26501191.
- Huynh AS, Abrahams DF, Torres MS, Baldwin MK, Gillies RJ, and Morse DL. Development of an orthotopic human pancreatic cancer xenograft model using ultrasound guided injection of cells. PLoS One 6: e20330, 2011. PMID: 21647423.
- Ideno N, Yamaguchi H, Okumura T, Huang J, Brun MJ, Ho ML, Suh J, Gupta S, Maitra A, and Ghosh B. A pipeline for rapidly generating genetically engineered mouse models of pancreatic cancer using in vivo CRISPR-Cas9-mediated somatic recombination. Lab Invest 99: 1233-1244, 2019. PMID: 30728464.
- Ijichi H, Chytil A, Gorska AE, Aakre ME, Fujitani Y, Fujitani S, Wright CV, and Moses HL. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev 20: 3147-3160, 2006. PMID: 17114585.
- Inkoom A, Ndemazie N, Affram K, Smith T, Zhu X, Underwood P, Krishnan S, Ofori E, Han B, Trevino J, and Agyare E. Enhancing efficacy of gemcitabine in pancreatic patient-derived xenograft mouse models. Int J Pharm X 2: 100056, 2020. PMID: 33015617.
- Izeradjene K, Combs C, Best M, Gopinathan A, Wagner A, Grady WM, Deng CX, Hruban RH, Adsay NV, Tuveson DA, and Hingorani SR. KrasG12D and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell 11: 229-243, 2007. PMID: 17349581.
- Izumchenko E, Paz K, Ciznadija D, Sloma I, Katz A, Vasquez-Dunddel D, Ben-Zvi I, Stebbing J, McGuire W, Harris W, Maki R, Gaya A, Bedi A, Zacharoulis S, Ravi R, Wexler LH, Hoque MO, Rodriguez-Galindo C, Pass H, Peled N, et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann Oncol 28: 2595-2605, 2017. PMID: 28945830.
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321: 1801-1806, 2008. PMID: 18772397.
- Kazi A, Xiang S, Yang H, Chen L, Kennedy P, Ayaz M, Fletcher S, Cummings C, Lawrence HR, Beato F, Kang Y, Kim MP, Delitto A, Underwood PW, Fleming JB, Trevino JG, Hamilton AD, and Sebti SM. Dual Farnesyl and Geranylgeranyl Transferase Inhibitor Thwarts Mutant KRAS-Driven Patient-Derived Pancreatic Tumors. Clin Cancer Res 25: 5984-5996, 2019. PMID: 31227505.
- Keng VW, Villanueva A, Chiang DY, Dupuy AJ, Ryan BJ, Matise I, Silverstein KA, Sarver A, Starr TK, Akagi K, Tessarollo L, Collier LS, Powers S, Lowe SW, Jenkins NA, Copeland NG, Llovet JM, and Largaespada DA. A conditional transposon-based insertional mutagenesis screen for genes associated with mouse hepatocellular carcinoma. Nat Biotechnol 27: 264-274, 2009. PMID: 19234449.
- Kogame A, Tagawa Y, Shibata S, Tojo H, Miyamoto M, Tohyama K, Kondo T, Prakash S, Shyu WC, and Asahi S. Pharmacokinetic and pharmacodynamic modeling of hedgehog inhibitor TAK-441 for the inhibition of Gli1 messenger RNA expression and antitumor efficacy in xenografted tumor model mice. Drug Metab Dispos 41: 727-734, 2013. PMID: 23298863.
- Kojima K, Vickers SM, Adsay NV, Jhala NC, Kim HG, Schoeb TR, Grizzle WE, and Klug CA. Inactivation of Smad4 accelerates KrasG12D-mediated pancreatic neoplasia. Cancer Res 67: 8121-8130, 2007. PMID: 17804724.
- Kong K, Guo M, Liu Y, and Zheng J. Progress in Animal Models of Pancreatic Ductal Adenocarcinoma. J Cancer 11: 1555-1567, 2020. PMID: 32047562.
- Kunnumakkara AB, Sung B, Ravindran J, Diagaradjane P, Deorukhkar A, Dey S, Koca C, Tong Z, Gelovani JG, Guha S, Krishnan S, and Aggarwal BB. Zyflamend suppresses growth and sensitizes human pancreatic tumors to gemcitabine in an orthotopic mouse model through modulation of multiple targets. Int J Cancer 131: E292-303, 2012. PMID: 21935918.
- Lau AN, Li Z, Danai LV, Westermark AM, Darnell AM, Ferreira R, Gocheva V, Sivanand S, Lien EC, Sapp KM, Mayers JR, Biffi G, Chin CR, Davidson SM, Tuveson DA, Jacks T, Matheson NJ, Yilmaz O, and Vander Heiden MG. Dissecting cell-type-specific metabolism in pancreatic ductal adenocarcinoma. Elife 9:e56782 2020. PMID: 32648540.
- Leach SD. Mouse models of pancreatic cancer: the fur is finally flying! Cancer Cell 5: 7-11, 2004. PMID: 14749121.
- Lesina M. "Charakterisierung der Funktion des Transkriptionsfaktors RelA/p65 im Ela-TGFα transgenen Mausmodell der pankreatischen Karzinogenese. 2013.
- Liang Q, Kong J, Stalker J, and Bradley A. Chromosomal mobilization and reintegration of Sleeping Beauty and PiggyBac transposons. Genesis 47: 404-408, 2009. PMID: 19391106.
- Luo D, Carter KA, Molins EAG, Straubinger NL, Geng J, Shao S, Jusko WJ, Straubinger RM, and Lovell JF. Pharmacokinetics and pharmacodynamics of liposomal chemophototherapy with short drug-light intervals. J Control Release 297: 39-47, 2019. PMID: 30684512.
- Luo D, Digiovanni MG, Wei R, Lacomb JF, Williams JL, Rigas B, and Mackenzie GG. Phospho-valproic acid (MDC-1112) reduces pancreatic cancer growth in patient-derived tumor xenografts and KPC mice: enhanced efficacy when combined with gemcitabine. Carcinogenesis 41: 927-939, 2020. PMID: 31584613.
- Lupo F, Piro G, Torroni L, Delfino P, Trovato R, Rusev B, Fiore A, Filippini D, De Sanctis F, Manfredi M, Marengo E, Lawlor RT, Martini M, Tortora G, Ugel S, Corbo V, Melisi D, and Carbone C. Organoid-Transplant Model Systems to Study the Effects of Obesity on the Pancreatic Carcinogenesis in vivo. Front Cell Dev Biol 8: 308, 2020. PMID: 32411709.
- Machinaga A, Hori Y, Shimizu K, Okahara K, Yanagita E, Miyoshi M, Itoh T, and Sasai K. Xenografts Derived From Patients' Ascites Recapitulate the Gemcitabine Resistance Observed in Pancreatic Cancer Patients. Pancreas 48: 1294-1302, 2019. PMID: 31688592.
- Maddipati R, and Stanger BZ. Pancreatic Cancer Metastases Harbor Evidence of Polyclonality. Cancer Discov 5: 1086-1097, 2015. PMID: 26209539.
- Maniati E, Bossard M, Cook N, Candido JB, Emami-Shahri N, Nedospasov SA, Balkwill FR, Tuveson DA, and Hagemann T. Crosstalk between the canonical NF-κB and Notch signaling pathways inhibits Pparγ expression and promotes pancreatic cancer progression in mice. Journal of Clinical Investigation 121: 4685-4699, 2011. PMID: 22056382.
- Maresch R, Mueller S, Veltkamp C, Ollinger R, Friedrich M, Heid I, Steiger K, Weber J, Engleitner T, Barenboim M, Klein S, Louzada S, Banerjee R, Strong A, Stauber T, Gross N, Geumann U, Lange S, Ringelhan M, Varela I, et al. Multiplexed pancreatic genome engineering and cancer induction by transfection-based CRISPR/Cas9 delivery in mice. Nat Commun 7: 10770, 2016. PMID: 26916719.
- Mato-Berciano A, Raimondi G, Maliandi MV, Alemany R, Montoliu L, and Fillat C. A NOTCH-sensitive uPAR-regulated oncolytic adenovirus effectively suppresses pancreatic tumor growth and triggers synergistic anticancer effects with gemcitabine and nab-paclitaxel. Oncotarget 8: 22700-22715, 2017. PMID: 28186974.
- Mazur PK, Einwachter H, Lee M, Sipos B, Nakhai H, Rad R, Zimber-Strobl U, Strobl LJ, Radtke F, Kloppel G, Schmid RM, and Siveke JT. Notch2 is required for progression of pancreatic intraepithelial neoplasia and development of pancreatic ductal adenocarcinoma. Proceedings of the National Academy of Sciences 107: 13438-13443, 2010. PMID: 20624967.
- Mazur PK, and Siveke JT. Genetically engineered mouse models of pancreatic cancer: unravelling tumour biology and progressing translational oncology. Gut 61: 1488-1500, 2012. PMID: 21873467.
- McNally LR, Welch DR, Beck BH, Stafford LJ, Long JW, Sellers JC, Huang ZQ, Grizzle WE, Stockard CR, Nash KT, and Buchsbaum DJ. KISS1 over-expression suppresses metastasis of pancreatic adenocarcinoma in a xenograft mouse model. Clin Exp Metastasis 27: 591-600, 2010. PMID: 20844932.
- Mesmar F, Dai B, Ibrahim A, Hases L, Jafferali MH, Jose Augustine J, DiLorenzo S, Kang Y, Zhao Y, Wang J, Kim M, Lin CY, Berkenstam A, Fleming J, and Williams C. Clinical candidate and genistein analogue AXP107-11 has chemoenhancing functions in pancreatic adenocarcinoma through G protein-coupled estrogen receptor signaling. Cancer Med 8: 7705-7719, 2019. PMID: 31568691.
- Milazzo FM, Vesci L, Anastasi AM, Chiapparino C, Rosi A, Giannini G, Taddei M, Cini E, Faltoni V, Petricci E, Battistuzzi G, Salvini L, Carollo V, and De Santis R. ErbB2 Targeted Epigenetic Modulation: Anti-tumor Efficacy of the ADC Trastuzumab-HDACi ST8176AA1. Front Oncol 9: 1534, 2019. PMID: 32039017.
- Moroz A, Wang YH, Sharib JM, Wei J, Zhao N, Huang Y, Chen Z, Martinko AJ, Zhuo J, Lim SA, Zhang LH, Seo Y, Carlin S, Leung KK, Collisson EA, Kirkwood KS, Wells JA, and Evans MJ. Theranostic Targeting of CUB Domain Containing Protein 1 (CDCP1) in Pancreatic Cancer. Clin Cancer Res 26: 3608-3615, 2020. PMID: 32341034.
- Morton JP, Jamieson NB, Karim SA, Athineos D, Ridgway RA, Nixon C, McKay CJ, Carter R, Brunton VG, Frame MC, Ashworth A, Oien KA, Evans TRJ, and Sansom OJ. LKB1 Haploinsufficiency Cooperates With Kras to Promote Pancreatic Cancer Through Suppression of p21-Dependent Growth Arrest. Gastroenterology 139: 586-597.e586, 2010. PMID: 20452353.
- Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, Jamieson NB, Oien KA, Lowy AM, Brunton VG, Frame MC, Evans TR, and Sansom OJ. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci U S A 107: 246-251, 2010. PMID: 20018721.
- Mottini C, Tomihara H, Carrella D, Lamolinara A, Iezzi M, Huang JK, Amoreo CA, Buglioni S, Manni I, Robinson FS, Minelli R, Kang Y, Fleming JB, Kim MP, Bristow CA, Trisciuoglio D, Iuliano A, Del Bufalo D, Di Bernardo D, Melisi D, et al. Predictive Signatures Inform the Effective Repurposing of Decitabine to Treat KRAS-Dependent Pancreatic Ductal Adenocarcinoma. Cancer Res 79: 5612-5625, 2019. PMID: 31492820.
- Mouhid L, Gomez de Cedron M, Garcia-Carrascosa E, Reglero G, Fornari T, and Ramirez de Molina A. Yarrow supercritical extract exerts antitumoral properties by targeting lipid metabolism in pancreatic cancer. PLoS One 14: e0214294, 2019. PMID: 30913248.
- Nikfarjam M, Yeo D, He H, Baldwin G, Fifis T, Costa P, Tan B, Yang E, Wen S, and Christophi C. Comparison of two syngeneic orthotopic murine models of pancreatic adenocarcinoma. J Invest Surg 26: 352-359, 2013. PMID: 23957638.
- Niknafs N, Zhong Y, Moral JA, Zhang L, Shao MX, Lo A, Makohon-Moore A, Iacobuzio-Donahue CA, and Karchin R. Characterization of genetic subclonal evolution in pancreatic cancer mouse models. Nat Commun 10: 5435, 2019. PMID: 31780749.
- Nishigaki T, Takahashi T, Serada S, Fujimoto M, Ohkawara T, Hara H, Sugase T, Otsuru T, Saito Y, Tsujii S, Nomura T, Tanaka K, Miyazaki Y, Makino T, Kurokawa Y, Nakajima K, Eguchi H, Yamasaki M, Mori M, Doki Y, et al. Anti-glypican-1 antibody-drug conjugate is a potential therapy against pancreatic cancer. Br J Cancer 122: 1333-1341, 2020. PMID: 32152502.
- Nolan-Stevaux O, Lau J, Truitt ML, Chu GC, Hebrok M, Fernandez-Zapico ME, and Hanahan D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev 23: 24-36, 2009. PMID: 19136624.
- Nosacka RL, Delitto AE, Delitto D, Patel R, Judge SM, Trevino JG, and Judge AR. Distinct cachexia profiles in response to human pancreatic tumours in mouse limb and respiratory muscle. J Cachexia Sarcopenia Muscle 11: 820-837, 2020. PMID: 32039571.
- Pan CH, Otsuka Y, Sridharan B, Woo M, Leiton CV, Babu S, Torrente Goncalves M, Kawalerski RR, JD KB, Chang DK, Biankin AV, Scampavia L, Spicer T, Escobar-Hoyos LF, and Shroyer KR. An unbiased high-throughput drug screen reveals a potential therapeutic vulnerability in the most lethal molecular subtype of pancreatic cancer. Mol Oncol 14: 1800-1816, 2020. PMID: 32533886.
- Partecke IL, Kaeding A, Sendler M, Albers N, Kuhn JP, Speerforck S, Roese S, Seubert F, Diedrich S, Kuehn S, Weiss UF, Mayerle J, Lerch MM, Hadlich S, Hosten N, Heidecke CD, Puls R, and von Bernstorff W. In vivo imaging of pancreatic tumours and liver metastases using 7 Tesla MRI in a murine orthotopic pancreatic cancer model and a liver metastases model. BMC Cancer 11: 40, 2011. PMID: 21276229.
- Perez-Mancera PA, Rust AG, van der Weyden L, Kristiansen G, Li A, Sarver AL, Silverstein KA, Grutzmann R, Aust D, Rummele P, Knosel T, Herd C, Stemple DL, Kettleborough R, Brosnan JA, Li A, Morgan R, Knight S, Yu J, Stegeman S, et al. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 486: 266-270, 2012. PMID: 22699621.
- Principe DR, DeCant B, Mascarinas E, Wayne EA, Diaz AM, Akagi N, Hwang R, Pasche B, Dawson DW, Fang D, Bentrem DJ, Munshi HG, Jung B, and Grippo PJ. TGFbeta Signaling in the Pancreatic Tumor Microenvironment Promotes Fibrosis and Immune Evasion to Facilitate Tumorigenesis. Cancer Res 76: 2525-2539, 2016. PMID: 26980767.
- Qiu W, and Su GH. Challenges and advances in mouse modeling for human pancreatic tumorigenesis and metastasis. Cancer Metastasis Rev 32: 83-107, 2013. PMID: 23114842.
- Qiu W, and Su GH. Development of orthotopic pancreatic tumor mouse models. Methods Mol Biol 980: 215-223, 2013. PMID: 23359156.
- Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, and Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature 456: 593-598, 2008. PMID: 19052619.
- Rad R, Rad L, Wang W, Cadinanos J, Vassiliou G, Rice S, Campos LS, Yusa K, Banerjee R, Li MA, de la Rosa J, Strong A, Lu D, Ellis P, Conte N, Yang FT, Liu P, and Bradley A. PiggyBac transposon mutagenesis: a tool for cancer gene discovery in mice. Science 330: 1104-1107, 2010. PMID: 20947725.
- Rad R, Rad L, Wang W, Strong A, Ponstingl H, Bronner IF, Mayho M, Steiger K, Weber J, Hieber M, Veltkamp C, Eser S, Geumann U, Ollinger R, Zukowska M, Barenboim M, Maresch R, Cadinanos J, Friedrich M, Varela I, et al. A conditional piggyBac transposition system for genetic screening in mice identifies oncogenic networks in pancreatic cancer. Nat Genet 47: 47-56, 2015. PMID: 25485836.
- Raimondi G, Mato-Berciano A, Pascual-Sabater S, Rovira-Rigau M, Cuatrecasas M, Fondevila C, Sanchez-Cabus S, Begthel H, Boj SF, Clevers H, and Fillat C. Patient-derived pancreatic tumour organoids identify therapeutic responses to oncolytic adenoviruses. EBioMedicine 56: 102786, 2020. PMID: 32460166.
- Rajeshkumar NV, Dutta P, Yabuuchi S, de Wilde RF, Martinez GV, Le A, Kamphorst JJ, Rabinowitz JD, Jain SK, Hidalgo M, Dang CV, Gillies RJ, and Maitra A. Therapeutic Targeting of the Warburg Effect in Pancreatic Cancer Relies on an Absence of p53 Function. Cancer Res 75: 3355-3364, 2015. PMID: 26113084.
- Roche S, O'Neill F, Murphy J, Swan N, Meiller J, Conlon NT, Geoghegan J, Conlon K, McDermott R, Rahman R, Toomey S, Straubinger NL, Straubinger RM, O'Connor R, McVey G, Moriarty M, and Clynes M. Establishment and Characterisation by Expression Microarray of Patient-Derived Xenograft Panel of Human Pancreatic Adenocarcinoma Patients. Int J Mol Sci 21: 2020. PMID: 32024004.
- Romero-Calvo I, Weber CR, Ray M, Brown M, Kirby K, Nandi RK, Long TM, Sparrow SM, Ugolkov A, Qiang W, Zhang Y, Brunetti T, Kindler H, Segal JP, Rzhetsky A, Mazar AP, Buschmann MM, Weichselbaum R, Roggin K, and White KP. Human Organoids Share Structural and Genetic Features with Primary Pancreatic Adenocarcinoma Tumors. Mol Cancer Res 17: 70-83, 2019. PMID: 30171177.
- Rowley M, Ohashi A, Mondal G, Mills L, Yang L, Zhang L, Sundsbak R, Shapiro V, Muders MH, Smyrk T, and Couch FJ. Inactivation of Brca2 promotes Trp53-associated but inhibits KrasG12D-dependent pancreatic cancer development in mice. Gastroenterology 140: 1303-1313 e1301-1303, 2011. PMID: 21199651.
- Russell J, Grkovski M, O'Donoghue IJ, Kalidindi TM, Pillarsetty N, Burnazi EM, Kulick A, Bahr A, Chang Q, LeKaye HC, de Stanchina E, Yu KH, and Humm JL. Predicting Gemcitabine Delivery by 18F-FAC PET in Murine Models of Pancreatic Cancer. J Nucl Med 62: 195-200, 2021. PMID: 32646874.
- Salvador-Barbero B, Alvarez-Fernandez M, Zapatero-Solana E, El Bakkali A, Menendez MDC, Lopez-Casas PP, Di Domenico T, Xie T, VanArsdale T, Shields DJ, Hidalgo M, and Malumbres M. CDK4/6 inhibitors impair recovery from cytotoxic chemotherapy in pancreatic adenocarcinoma. Cancer Cell 37: 340-353 2020. PMID: 32109375.
- Sandgren E. P. LNC, Qiu T.H., Palmiter R.D., Brinster R.L., Lee D.C. Transforming Growth Factor Alpha Dramatically Enhances Oncogene-Induced Carcinogenesis in Transgenic Mouse Pancreas and Liver. Mol Cell Biol 320-330, 1993. PMID: 8417334.
- Sandgren EP, Luetteke NC, Palmiter RD, Brinster RL, and Lee DC. Overexpression of TGF alpha in transgenic mice: induction of epithelial hyperplasia, pancreatic metaplasia, and carcinoma of the breast. Cell 61: 1121-1135, 1990. PMID: 1693546.
- Schonhuber N, Seidler B, Schuck K, Veltkamp C, Schachtler C, Zukowska M, Eser S, Feyerabend TB, Paul MC, Eser P, Klein S, Lowy AM, Banerjee R, Yang F, Lee CL, Moding EJ, Kirsch DG, Scheideler A, Alessi DR, Varela I, et al. A next-generation dual-recombinase system for time- and host-specific targeting of pancreatic cancer. Nat Med 20: 1340-1347, 2014. PMID: 25326799.
- Shakya R, Reid LJ, Reczek CR, Cole F, Egli D, Lin CS, deRooij DG, Hirsch S, Ravi K, Hicks JB, Szabolcs M, Jasin M, Baer R, and Ludwig T. BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science 334: 525-528, 2011. PMID: 22034435.
- Siveke JT, Einwachter H, Sipos B, Lubeseder-Martellato C, Kloppel G, and Schmid RM. Concomitant pancreatic activation of KrasG12D and Tgfa results in cystic papillary neoplasms reminiscent of human IPMN. Cancer Cell 12: 266-279, 2007. PMID: 17785207.
- Skoulidis F, Cassidy LD, Pisupati V, Jonasson JG, Bjarnason H, Eyfjord JE, Karreth FA, Lim M, Barber LM, Clatworthy SA, Davies SE, Olive KP, Tuveson DA, and Venkitaraman AR. Germline Brca2 heterozygosity promotes KrasG12D -driven carcinogenesis in a murine model of familial pancreatic cancer. Cancer Cell 18: 499-509, 2010. PMID: 21056012.
- Solanki A, King D, Thibault G, Wang L, and Gibbs SL. Quantification of fluorophore distribution and therapeutic response in matched in vivo and ex vivo pancreatic cancer model systems. PLoS One 15: e0229407, 2020. PMID: 32097436.
- Stuelten CH, Barbul A, Busch JI, Sutton E, Katz R, Sato M, Wakefield LM, Roberts AB, and Niederhuber JE. Acute wounds accelerate tumorigenesis by a T cell-dependent mechanism. Cancer Res 68: 7278-7282, 2008. PMID: 18794114.
- Sun Y, He W, Luo M, Zhou Y, Chang G, Ren W, Wu K, Li X, Shen J, Zhao X, and Hu Y. SREBP1 regulates tumorigenesis and prognosis of pancreatic cancer through targeting lipid metabolism. Tumour Biol 36: 4133-4141, 2015. PMID: 25589463.
- Tadros S, Shukla SK, King RJ, Gunda V, Vernucci E, Abrego J, Chaika NV, Yu F, Lazenby AJ, Berim L, Grem J, Sasson AR, and Singh PK. De Novo Lipid Synthesis Facilitates Gemcitabine Resistance through Endoplasmic Reticulum Stress in Pancreatic Cancer. Cancer Res 77: 5503-5517, 2017. PMID: 28811332.
- Tanaka R, Kageyama K, Kimura K, Eguchi S, Tauchi J, Shinkawa H, Ohira GO, Yamazoe S, Yamamoto A, Tanaka S, Amano R, Tanaka H, Yashiro M, Kubo S, and Ohira M. Establishment of a Liver Transplant Patient-derived Tumor Xenograft (PDX) Model Using Cryopreserved Pancreatic Ductal Adenocarcinoma. Anticancer Res 40: 2637-2644, 2020. PMID: 32366408.
- Tinder TL, Subramani DB, Basu GD, Bradley JM, Schettini J, Million A, Skaar T, and Mukherjee P. MUC1 enhances tumor progression and contributes toward immunosuppression in a mouse model of spontaneous pancreatic adenocarcinoma. J Immunol 181: 3116-3125, 2008. PMID: 18713982.
- Torres C, Mancinelli G, Cordoba-Chacon J, Viswakarma N, Castellanos K, Grimaldo S, Kumar S, Principe D, Dorman MJ, McKinney R, Hirsch E, Dawson D, Munshi HG, Rana A, and Grippo PJ. p110γ deficiency protects against pancreatic carcinogenesis yet predisposes to diet-induced hepatotoxicity. Proc Natl Acad Sci U S A 116: 14724-14733, 2019. PMID: 31266893.
- Trevino JG, Summy JM, Lesslie DP, Parikh NU, Hong DS, Lee FY, Donato NJ, Abbruzzese JL, Baker CH, and Gallick GE. Inhibition of SRC expression and activity inhibits tumor progression and metastasis of human pancreatic adenocarcinoma cells in an orthotopic nude mouse model. Am J Pathol 168: 962-972, 2006. PMID: 16507911.
- Tseng WW, Winer D, Kenkel JA, Choi O, Shain AH, Pollack JR, French R, Lowy AM, and Engleman EG. Development of an orthotopic model of invasive pancreatic cancer in an immunocompetent murine host. Clin Cancer Res 16: 3684-3695, 2010. PMID: 20534740.
- Tuveson DA, Zhu L, Gopinathan A, Willis NA, Kachatrian L, Grochow R, Pin CL, Mitin NY, Taparowsky EJ, Gimotty PA, Hruban RH, Jacks T, and Konieczny SF. Mist1-KrasG12D knock-in mice develop mixed differentiation metastatic exocrine pancreatic carcinoma and hepatocellular carcinoma. Cancer Res 66: 242-247, 2006. PMID: 16397237.
- Underwood PW, Zhang DY, Cameron ME, Gerber MH, Delitto D, Maduka MU, Cooper KJ, Han S, Hughes SJ, Judge SM, Judge AR, and Trevino JG. Nicotine Induces IL-8 Secretion from Pancreatic Cancer Stroma and Worsens Cancer-Induced Cachexia. Cancers (Basel) 12: 2020. PMID: 32024069.
- Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernandez-Mateos J, Khan K, Lampis A, Eason K, Huntingford I, Burke R, Rata M, Koh DM, Tunariu N, Collins D, Hulkki-Wilson S, Ragulan C, Spiteri I, Moorcraft SY, Chau I, Rao S, et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 359: 920-926, 2018. PMID: 29472484.
- Wagner M, Greten FR, Weber CK, Koschnick S, Mattfeldt T, Deppert W, Kern H, Adler G, and Schmid RM. A murine tumor progression model for pancreatic cancer recapitulating the genetic alterations of the human disease. Genes Dev 15: 286-293, 2001. PMID: 11159909.
- Wagner M, Luhrs H, Kloppel G, Adler G, and Schmid RM. Malignant transformation of duct-like cells originating from acini in transforming growth factor transgenic mice. Gastroenterology 115: 1254-1262, 1998. PMID: 9797382.
- Walsh AJ, Castellanos JA, Nagathihalli NS, Merchant NB, and Skala MC. Optical Imaging of Drug-Induced Metabolism Changes in Murine and Human Pancreatic Cancer Organoids Reveals Heterogeneous Drug Response. Pancreas 45: 863-869, 2016. PMID: 26495796.
- Wang G, Zhou Z, Zhao Z, Li Q, Wu Y, Yan S, Shen Y, and Huang P. Enzyme-Triggered Transcytosis of Dendrimer-Drug Conjugate for Deep Penetration into Pancreatic Tumors. ACS Nano 14: 4890-4904, 2020. PMID: 32286784.
- Wang J, Chan DKW, Sen A, Ma WW, and Straubinger RM. Tumor Priming by SMO Inhibition Enhances Antibody Delivery and Efficacy in a Pancreatic Ductal Adenocarcinoma Model. Mol Cancer Ther 18: 2074-2084, 2019. PMID: 31363010.
- Wang Y, Park JYP, Pacis A, Denroche RE, Jang GH, Zhang A, Cuggia A, Domecq C, Monlong J, Raitses-Gurevich M, Grant RC, Borgida A, Holter S, Stossel C, Bu S, Masoomian M, Lungu IM, Bartlett JMS, Wilson JM, Gao ZH, et al. A Preclinical Trial and Molecularly Annotated Patient Cohort Identify Predictive Biomarkers in Homologous Recombination-deficient Pancreatic Cancer. Clin Cancer Res 26: 5462-5476, 2020. PMID: 32816949.
- Weber J, Braun CJ, Saur D, and Rad R. In vivo functional screening for systems-level integrative cancer genomics. Nat Rev Cancer 20: 573-593, 2020. PMID: 3263489.
- Wen CL, Huang K, Jiang LL, Lu XX, Dai YT, Shi MM, Tang XM, Wang QB, Zhang XD, Wang PH, Li HT, Ruan XX, Wang LW, Wang XJ, Wang Q, Lu W, Xiang XQ, Sun X, Xu YH, Lai LH, et al. An allosteric PGAM1 inhibitor effectively suppresses pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A 116: 23264-23273, 2019. PMID: 31662475.
- Weng CC, Hawse JR, Subramaniam M, Chang VHS, Yu WCY, Hung WC, Chen LT, and Cheng KH. KLF10 loss in the pancreas provokes activation of SDF-1 and induces distant metastases of pancreatic ductal adenocarcinoma in the KrasG12D p53flox/flox model. Oncogene 36: 5532-5543, 2017. PMID: 28581520.
- Weng CC, Hsieh MJ, Wu CC, Lin YC, Shan YS, Hung WC, Chen LT, and Cheng KH. Loss of the transcriptional repressor TGIF1 results in enhanced Kras-driven development of pancreatic cancer. Mol Cancer 18: 96, 2019. PMID: 31109321.
- Xu W, Yang XW, Zhao ZY, Dong B, Guan XY, Tian XY, Qian HG, and Hao CY. Establishment of pancreatic cancer patient-derived xenograft models and comparison of the differences among the generations. Am J Transl Res 11: 3128-3139, 2019. PMID: 31217882.
- Yang D, Zhang Q, Ma Y, Che Z, Zhang W, Wu M, Wu L, Liu F, Chu Y, Xu W, McGrath M, Song C, and Liu J. Augmenting the therapeutic efficacy of adenosine against pancreatic cancer by switching the Akt/p21-dependent senescence to apoptosis. EBioMedicine47: 114-127, 2019. PMID: 31495718.
- Yang H, Tong Z, Shen L, Sun YU, Hoffman RM, and Huang J. Brucea javanica Increases Survival and Enhances Gemcitabine Efficacy in a Patient-derived Orthotopic Xenograft (PDOX) Mouse Model of Pancreatic Cancer. Anticancer Res 40: 4969-4978, 2020. PMID: 32878785.
- Yao Y, Yao Q, Fu Y, Tian X, An Q, Yang L, Su H, Lu W, Hao C, and Zhou T. Pharmacokinetic/Pharmacodynamic Modeling of the Anti-Cancer Effect of Dexamethasone in Pancreatic Cancer Xenografts and Anticipation of Human Efficacious Doses. J Pharm Sci 109: 1169-1177, 2020. PMID: 31655033.
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, Yan H, Wang W, Chen S, Viale A, Zheng H, Paik JH, Lim C, Guimaraes AR, Martin ES, Chang J, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149: 656-670, 2012. PMID: 22541435.
- Yusa K, Rad R, Takeda J, and Bradley A. Generation of transgene-free induced pluripotent mouse stem cells by the piggyBac transposon. Nat Methods 6: 363-369, 2009. PMID: 19337237.
- Zhou C, Qian W, Li J, Ma J, Chen X, Jiang Z, Cheng L, Duan W, Wang Z, Wu Z, Ma Q, and Li X. High glucose microenvironment accelerates tumor growth via SREBP1-autophagy axis in pancreatic cancer. J Exp Clin Cancer Res 38: 302, 2019. PMID: 31296258.