
Entry Version:
Citation:
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2020.12
| Attachment | Size |
|---|---|
| 971.74 KB | |
| 1.2 MB |
I. Introduction
Intracellular Ca2+ signaling and the primary function of the exocrine pancreas to secrete digestive enzymes, are intimately linked. A secretagogue-induced rise in the level of intracellular Ca2+ signaling is the pivotal intracellular event triggering both the molecular machinery that results in exocytosis of zymogen granules and also the activation of ion channels that lead to the secretion of the primary fluid from acinar cells. Given the need to ensure vectoral secretion of a potentially hazardous cargo into the lumen of the gland, the Ca2+ signals and activation of effectors are elaborately orchestrated in time and space. The importance of the physiological Ca2+ signal is reinforced by increasing evidence that disordered Ca2+ signaling is causally associated with pancreatic pathology (45). The aim of this chapter is two-fold; first to provide the reader with an overview of the spatiotemporal features of intracellular Ca2+ signals in acinar cells as measured by contemporary high-resolution fluorescence measurements. Secondly, a review will be presented of the current understanding of the molecular mechanisms underpinning Ca2+ release, Ca2+ influx and clearance; processes that are fundamental for the specific properties of intracellular Ca2+ signals in acinar cells. The reader is referred to various “Method” submissions in The Pancreapedia regarding current imaging techniques used to monitor intracellular Ca2+ changes with high spatial and temporal resolution in acinar cells.
A. Signal Transduction Leading to Secretagogue-Induced Cytosolic Calcium Signals in Acinar Cells
The primary pancreatic secretagogues Cholecystokinin (CCK), Acetylcholine (ACh) and Bombesin share a common general mechanism to raise [Ca2+]i. It is well established that each agonist binds a specific, cognate, 7-transmembrane domain receptor in the plasma membrane (PM) that couples to the heterotrimeric G protein, Gαq. Activation of Gαq results in the increased activity of phosphoinositide (PI)- specific phospholipase C which cleaves phosphatidylinositol, 4,5-bisphosphate (PIP2) in the PM and subsequently generates the second messengers inositol 1,4,5-trisphosphate (IP3) and diacylglycerol. Historically, studies in pancreatic acinar cells have played important roles in establishing the signal transduction pathway utilized by so-called “Calcium Mobilizing” agents. A seminal paper by Streb and colleagues (137) was the first to demonstrate that IP3 could induce Ca2+ release in studies using permeabilized pancreatic acini (137). Notably, release could only be evoked from rough ER vesicles but not mitochondria or vesicles prepared from plasma membrane (135). These data confirmed earlier studies that secretagogues stimulation resulted in substantial Ca2+ loss from ER but not zymogen granules or mitochondria (32). Finally, the causative “loop” was then closed by the demonstration that stimulation with pancreatic secretagogues resulted in the rapid formation of IP3 (136). Notably, this pathway has subsequently been shown to be ubiquitous and accounts for the generation of Ca2+ signaling events important for a multitude of cellular events in all tissue systems.
II. The Characteristics of Secretagogue- Induced Cytosolic Calcium Signals in Acinar Cells
A. Temporal Properties at High Concentrations of Secretagogues
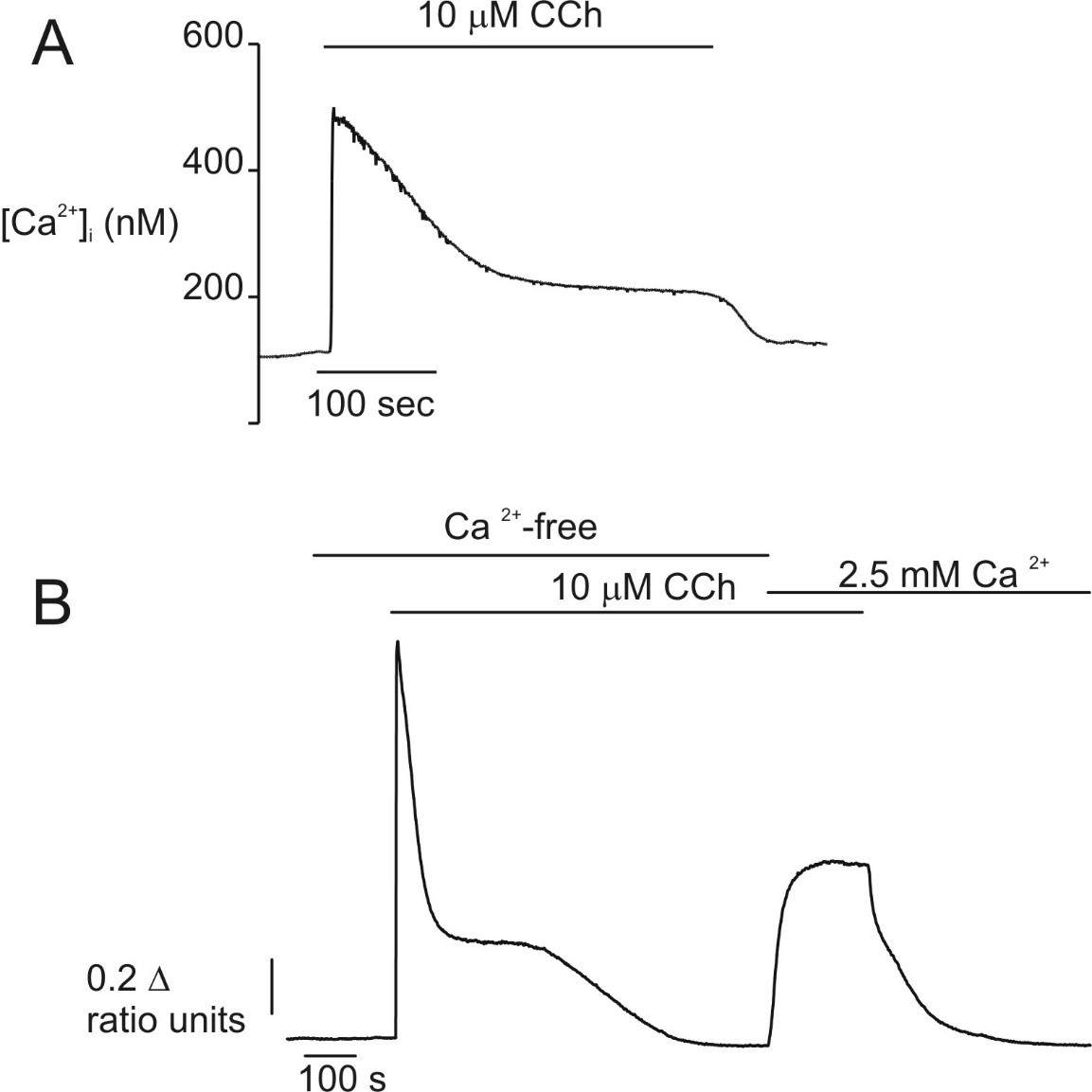
In common with many non-electrically excitable cell types, the spatiotemporal pattern of Ca2+ signals elicited in pancreatic acinar cells are dependent on the concentration of agonist. Regarding the temporal characteristics of signals in isolated pancreatic acinar cells, experiments monitoring the global intracellular Ca2+ concentration ([Ca2+]i) using fluorometric techniques in single rodent cells reported that maximum concentrations of CCK, ACh or bombesin evoked a similar Ca2+ signal. Exposure to either agonist, results in a rapid 5-12 fold increase in [Ca2+]i from a basal value of [Ca2+] ~75-150 nM to reach a peak within seconds of approximately 1 μM (Figure 1A). This peak then declined over 2-5 minutes to reach a new plateau level around 100 nM above basal, which is maintained as long as the agonist is present (89, 118, 138, 177). The initial peak was shown to be the result of Ca2+ release from intracellular stores, since the early transient response was essentially unaffected by removal of extracellular Ca2+ while the later plateau phase was absent (Figure 1B) (107, 138, 152, 177). Removal of extracellular Ca2+ during the plateau phase resulted in the rapid attenuation of the signal (Figure 2B), thus indicating an absolute dependence on extracellular Ca2+ for this maintained phase and implying the presence of a mechanism for Ca2+ influx from the extracellular milieu. A study monitoring ACh-induced Ca2+ signals in human pancreatic acinar cells confirmed this general paradigm; the initial phase of the signal occurs independently of extracellular Ca2+, while the sustained plateau requires the presence of Ca2+ in the extracellular milieu (83).
Figure 1. [Ca2+]i changes evoked by maximal concentrations of secretagogues in single cells. Stimulation with maximal concentrations of secretagogues results in a characteristic “peak and plateau” type Ca2+ signal. A) In a single fura-2 loaded rat pancreatic acinar cell, stimulation with 10 μM CCh resulted in a sharp increase in [Ca2+]i which subsequently declines to a new plateau level which was maintained throughout the period of secretagogue application. (B) In the absence of extracellular Ca2+ the initial peak can be initiated, but is only transient indicating that the initial phase of the response is a result of Ca2+ release from intracellular stores. Readmission of extracellular Ca2+ restores the “plateau” phase of the response indicating that Ca2+ influx is required to maintain this portion of the response.
B. Temporal Properties at Physiological Concentrations of Secretagogues
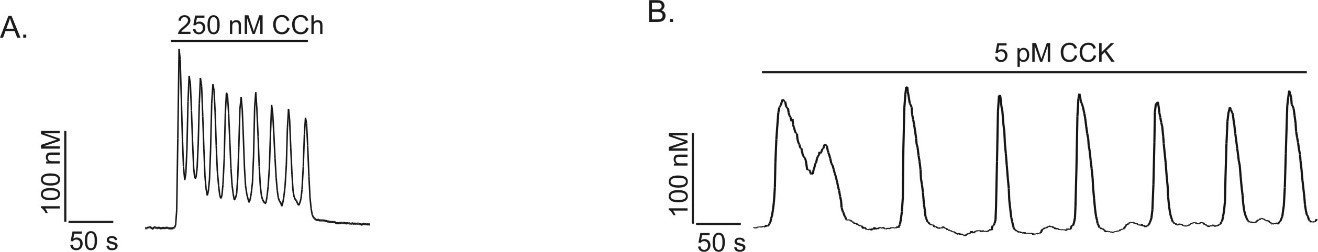
In contrast to the situation with maximal concentrations of agonists, similar experimental techniques demonstrated that lower, physiological concentrations of agonist resulted in the initiation of Ca2+ oscillations (118, 129, 138, 153, 177). These oscillations are characterized by repetitive, regular cycles of elevated and subsequently decreasing Ca2+ levels. In single acinar cells or small acini, physiological concentrations of secretagogues (1-50 pM CCK; 50-300 nM ACh) induce after a latency of 30s -2 min fairly regular Ca2+ oscillations at a frequency of between 1- 6 cycles per minute. The maximal global [Ca2+]i reached during the release phase is generally between 200 nM and 1 μM. At least in mouse pancreatic acinar cells the global temporal profile stimulated by ACh differs from that stimulated by CCK and bombesin (50, 72, 105, 179). Oscillations stimulated by peptide secretagogues tend to be characterized by slow, relatively long-lived transients originating and returning to basal levels between Ca2+ spikes while ACh-induced oscillations are characterized by faster short lasting transients originating from an elevated plateau (Figure 2A and B).
Figure 2. Physiological concentrations of agonists evoke Ca2+ oscillations. In single mouse pancreatic acinar cells low concentrations of secretagogue (50-400 nM ACh or 1-50 pM CCK) evoke repetitive Ca2+ transients termed Ca2+ oscillations. In small clusters of mouse acinar cells muscarinic receptor and CCK-stimulation resulted in distinct global temporal patterns of Ca2+ signal. (A) CCh-stimulation results in sinusoidal oscillations superimposed on an elevated baseline, while as shown in (B), CCK stimulation results in much less frequent, broader transients which originate and return to basal [Ca2+]i levels between transients. (From reference (133)).
C. Mechanisms Underlying Oscillating Ca2+ Signals
Consistent with a prominent role for receptor stimulated, Gαq-stimulated, PIP2 hydrolysis as the underlying mechanism, Ca2+ oscillations in pancreatic acinar cells can be mimicked by agents which activate heterotrimeric G proteins such as GTPγS, sodium fluoride or mastoparan and by introduction of IP3 into the cell for example via dialysis from a whole-cell patch clamp pipette (105, 161, 179). Oscillations are primarily the result of cycles of intracellular Ca2+ release and ATP dependent reuptake because the oscillations can be initiated in the absence of extracellular Ca2+ and are inhibited by agents, which deplete ATP or inhibit the Ca2+ pump on the endoplasmic reticulum (74, 153, 177). Although Ca2+ release during each cycle only minimally depletes the intracellular Ca2+ store (111) and reuptake is efficient (96), the maintenance of oscillatory behavior is dependent on extracellular Ca2+. These observations again indicate an absolute requirement for Ca2+ influx to sustain the Ca2+ signal presumably primarily by maintaining the level of Ca2+ in the intracellular store (177, 179).
Oscillating levels of IP3 are not necessary per se for oscillatory behavior, since non-metabolizable IP3 is capable of initiating Ca2+ oscillations in mouse pancreatic acinar cells (161). These data indicate that the mechanism underlying Ca2+ oscillations is most likely the result of an inherent property of the Ca2+ release mechanism. Nevertheless, these data do not preclude the possibility that upon agonist stimulation that the [IP3] itself fluctuates and contributes to the kinetics of Ca2+ release. Indeed mathematical predictions based on the experimental behavior of Ca2+ oscillations when Ca2+ or IP3 is artificially raised during agonist exposure predict that the [IP3] is itself oscillating during CCK stimulation (130). One mechanism whereby oscillating IP3 could occur is through periodic activation cycles of RGS proteins. RGS proteins stimulate the GTPase activity of Gα subunits thereby terminating the stimulus for activation of effectors such as PLC β. In rat pancreatic acinar cells infusion of RGS proteins via the patch-pipette results in dampening of Ca2+ signals (85, 171, 184, 185). Interestingly, the common catalytic core of RGS proteins, the so-called RGS box is much less effective than infusion of full-length RGS proteins (185). In addition, specific RGS proteins appear to affect the Ca2+ signals generated by different secretagogues differentially (171). These observations may indicate that individual RGS proteins are associated in a signaling complex with specific secretagogues receptors and other signaling proteins. This interaction may impact the kinetics of IP3 production and contribute to the agonist–specific characteristics of secretagogue-stimulated Ca2+ signals in pancreatic acinar cells.
D. Spatial Properties of Ca2+ Signals
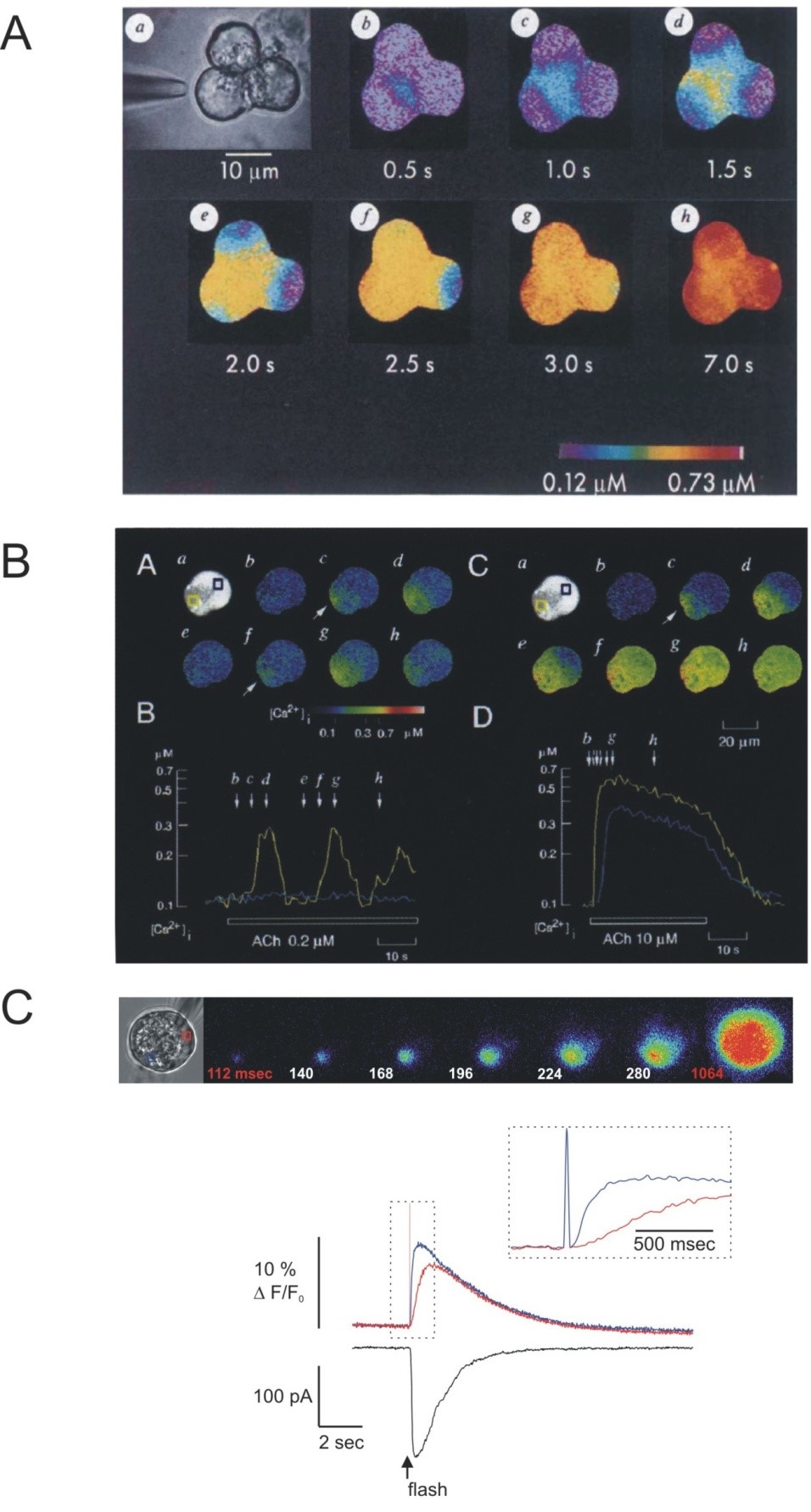
Measurements of global Ca2+ signals have provided a wealth of data regarding the general temporal properties of Ca2+ signals in pancreatic acinar cells. However, by the nature of the measurements, the experiments report the [Ca2+]i as a mean value integrated from throughout the cell. Modern imaging techniques however, allow the monitoring of [Ca2+]i at the subcellular level and in multiple cells of a coupled acinus. Probes, both chemical, and genetically encoded sensors are available with a choice of spectral characteristics and affinities for Ca2+ and with the ability to be targeted to various cellular compartments (24). Importantly, transgenic animals have been generated expressing a variety of these probes (104, 182). Highlighting the promise of these techniques to enable the study of Ca2+ signals in living animals and thus in their native environment, an initial study using intra vital two-photon microscopy in YC3.60 expressing animals reported Ca2+ signals following the injection of the muscarinic agonist bethanechol with characteristics similar to that seen in isolated acini (64). The combination of the flexibility of the probes plus the imaginative use of digital imaging techniques, such as confocal, multi-photon and total internal reflection fluorescence (TIRF) microscopy has revealed that the Ca2+ signal displays remarkable spatial intricacy, which appears to be fundamental for the appropriate activation of downstream effectors. An early study by Kasai and Augustine utilizing digital imaging of small acinar clusters demonstrated a profound spatial heterogeneity in the Ca2+ signal following stimulation with a maximal concentration of ACh (Figure 3A). Stimulation resulted in the initiation of Ca2+ release in the apical region of the cell immediately below the luminal plasma membrane and the subsequent spread of the signal as a wave towards the basal aspects of the cell (65). This Ca2+ wave has generally been reported to travel across the cell at a speed of between 5 ~ 45 μm/s, consistent with the velocity observed in other cell types (52, 53, 65, 101, 134, 150). These data were somewhat counterintuitive since contemporary studies had demonstrated that secretagogues receptors were expressed on the basolateral face of the cell (123) and it was known that the ER, the presumed site of Ca2+ release was present throughout the cell. Nathanson and colleagues, using line-scanning confocal microscopy later confirmed that a similar pattern of apical to basal Ca2+ wave was initiated by high concentrations of CCK (101). At physiological concentrations of agonists using similar techniques, Ca2+ signals are also shown to initiate in the apical region of acinar cells (66, 148). This initial site of Ca2+ release has been termed the “trigger-zone” (66) (Figure 3B). Ca2+ release invariably occurs at this specialized site even under conditions where stimulation of agonist is restricted to the basal region by focal application of agonist. This has been most elegantly demonstrated by focal flash photolysis of caged-carbachol contained in a whole cell patch clamp pipette isolated in the base of the cell (3). It should be noted however, that the apical portion of the acinus, immediately proximal to the tight junctions may express a relatively high number of secretagogue receptors since this area has been reported to be most sensitive to focal agonist stimulation (79, 126). At threshold concentrations of ACh, repetitive short lasting Ca2+ transients are initiated which strikingly are contained to the apical third of the cell and do not propagate to the basal region (66, 148). These spikes, although short lived have been shown using low-affinity Ca2+ indicators to be of large amplitude in the order of 1-4 μM Ca2+ (62). At intermediate concentrations of ACh, apically initiated global Ca2+ transients dominate, often superimposed on a slight global elevation of [Ca2+]i. The frequency of these transients corresponds to the frequency of oscillations noted in microfluorimetry studies. Low concentrations of CCK predominately result in apically initiated global Ca2+ signals, which are of longer duration (105, 179). In studies of whole cell patch-clamped acinar cells, where the Ca2+ buffering of the cell is set by dialysis from the patch-pipette, the broad CCK-induced transients have been reported to be preceded by short lasting apically localized transients (115). Studies indicate that the precise site of initiation of each transient by specific agonists is very similar, but possibly not identical (126). The site of initiation of each Ca2+ transient in the trigger zone is nevertheless tightly coupled functionally to both the exocytosis of zymogen granules and the activation of Cl- channels required for the process of fluid secretion from the pancreatic acinar cells (62, 110). Notably, the entire spectrum of signals evoked by secretagogues, from localized apical release to propagation of global Ca2+ waves can be mimicked by global, uniform application of IP3 either through the patch pipette or via flash photolysis from a caged precursor (Figure 3C) (38, 50, 134, 148). It is envisioned that future work utilizing intra vital microscopy in animals expressing genetically encoded Ca2+ indicators will further our understanding of the spatial and temporal properties of Ca2+ signals evoked by physiological stimulation.
Figure 3. Spatial characteristics of Ca2+ signals in pancreatic acini. Digital imaging of Ca2+ indicators reveals spatial homogeneity in agonist-stimulated Ca2+ signals. (A) Stimulation of a triplet of mouse pancreatic acinar cells (shown in a) with a maximal concentration of ACh results in the initiation of the Ca2+ signal in the extreme apical portion of the acinar cells (shown in b). The signal subsequently spread towards the basal aspects of each cell (From reference (65)). The Pseudocolor scale indicates the levels of [Ca2+]i. In (B), a single pancreatic acinar cell is stimulated with a threshold concentration of ACh. Ca2+ signals are again initiated in the apical portion of the cell but remain in the apical third of the cell without spreading to the basal aspects of the cell (images (B) Ab-Ah). The kinetic recorded from an apical region of interest (yellow trace in (B)) and from the basal region (blue trace) demonstrates that [Ca2+]i elevations are only observed in the apical pole of the acinar cell under these conditions (From reference (66)). (C) Shows the changes in Ca2+ following photolytic liberation of 1,4,5 InsP3 from a caged precursor induced into a single mouse acinar cell via a whole-cell patch clamp pipette. Following global elevation of 1,4,5-InsP3 Ca2+ changes initially occur at the apical pole of the acinar cell and spread to the basal pole in a similar fashion to secretagogue stimulation. The kinetic shows the Ca2+ changes in the apical (blue trace) vs. basal pole (red trace) of the cell together with the activation of a chloride conductance as measured by whole cell patch clamp. (From reference (50)).
E. The Impact of Cell-Cell Communication on Ca2+ Signaling
Individual cells in the pancreatic acinus are extensively coupled by the expression of gap junctional proteins (91). These channels effectively allow the passage of small molecules up to a molecular mass of 1-2 kDa between cells and furthermore provide electrical coupling of large numbers of cells in the acinus. While stimulation of small acini with maximal concentrations of agonist results in the Ca2+ dependent closure of these junctions (63, 180), at physiological levels of CCK and bombesin where global Ca2+ transients predominate the junctions remain open and Ca2+ signals appear to spread as waves between individual cells (131, 180). In contrast, brief, apically confined transients initiated by threshold concentrations of ACh have been reported not to propagate between coupled cells (180). Each cycle of CCK-induced intercellular Ca2+ signaling has been reported to be initiated by a “pacemaker cell” (180). This pacemaker presumably represents the individual cell within the acinus most sensitive to agonist. The propagation of a Ca2+ wave between adjacent cells obviously requires relatively long-range messengers. It appears that IP3 and small amounts of Ca2+ are capable of diffusing between coupled cells to act in concert in this manner providing a signal to synchronize the intercellular Ca2+ wave. The primary evidence for this contention is that a Ca2+ signal can be observed in neighboring cells when 1,4,5-IP3 is injected into an unstimulated individual cell. In addition, while Ca2+ injected into a resting cell fails to measurably increase [Ca2+]i in adjacent cells, microinjection of Ca2+ into cells previously stimulated with threshold concentrations of CCK leads to a measurable increase in [Ca2+]i in neighboring cells (180). These data are consistent with Ca2+ acting to facilitate further Ca2+ release from intracellular stores as will described in detail in the remainder of this chapter. The physiological function of propagating Ca2+ waves in pancreatic acinar cells is not at present firmly established. A reasonable proposal however, is that gap-junctional communication represents a mechanism to increase the responsiveness of an acinus to threshold concentrations of agonist. In this scenario, the acinus is rendered as sensitive to secretagogues stimulation as the pacemaker cell. In support of this idea, isolated single cells are much less sensitive to secretagogues stimulation than isolated acini and in addition experimental maneuvers which increase gap-junctional permeability lead to increased secretagogue induced amylase secretion (131).
III. Molecular Mechanisms Underlying Ca2+ Signaling in Pancreatic Acinar Cells
A. Intracellular Ca2+ Release
IP3-Induced Ca2+ Release
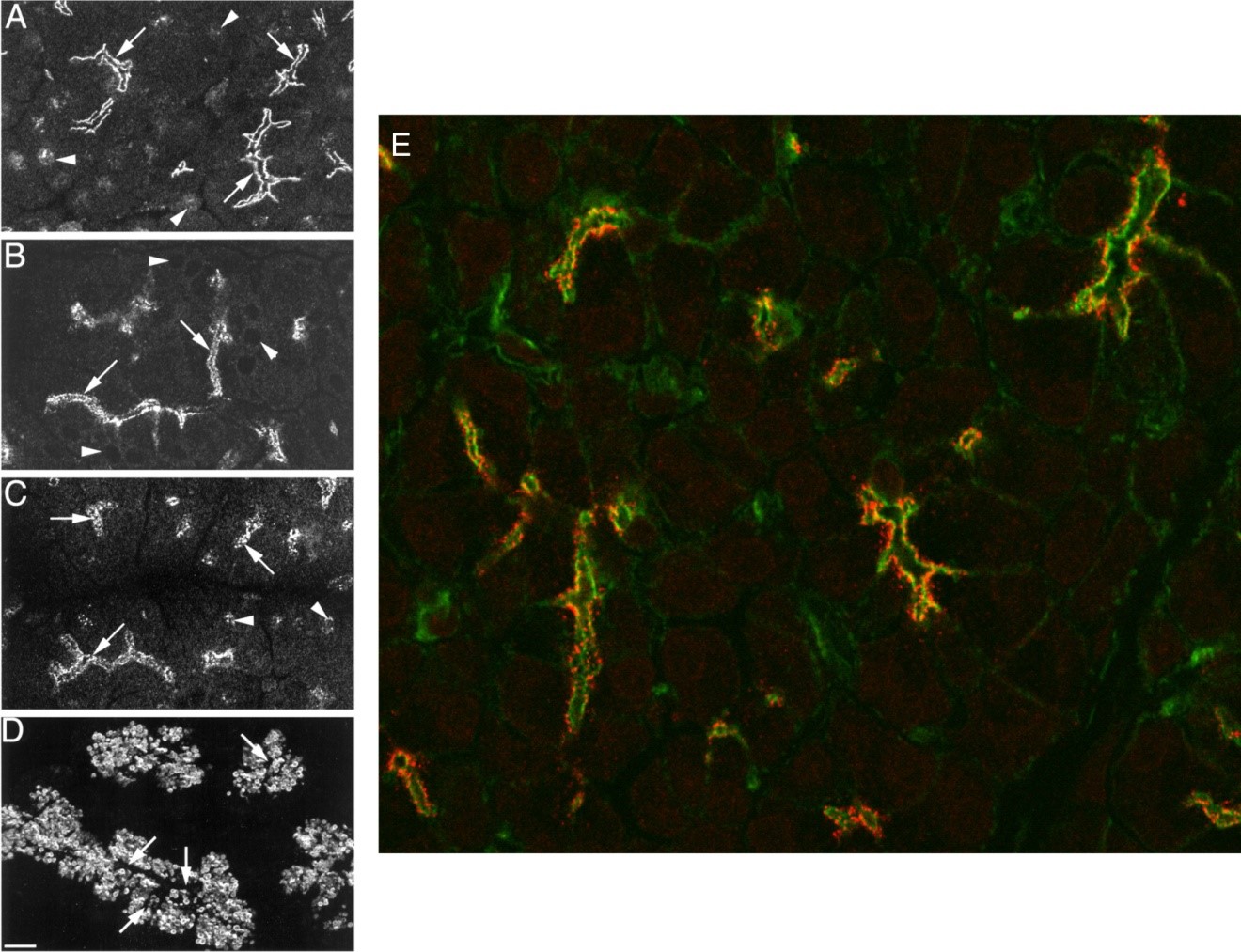
The invariable apical initiation of Ca2+ release dictates the view that this trigger zone must represent a specialized region of ER, highly sensitive to IP3. Studies have shown that this exquisite sensitivity to IP3 is a result of the abundant expression of IP3 receptors (IP3R) in the extreme apical region of pancreatic acini (75, 100, 176). IP3R were first isolated and cloned from cerebellum and have subsequently been shown to represent a family of three proteins named the type-1 IP3R (IP3R1), type-2 IP3R (IP3R2) and type-3 IP3R (IP3R3) which are all related to the ryanodine receptor Ca2+ release channel (41, 88, 94). Initially, it was reported that IP3R3 was expressed in the apical pole (100). Later studies however, showed that all three subtypes, had essentially identical expression; all IP3R were excluded from areas containing zymogen granules and were apparent immediately below the apical and lateral plasma membrane (75, 176) (Figure 4A-D). This localization is essentially identical to the “terminal-web” of actin based cytoskeleton in this region (Figure 4E). The localization to this region may be dependent on lipid rafts because cholesterol depletion results in the redistribution of IP3R and disruption of the apical to basal Ca2+ wave (99). By this technique no other significant localization of IP3R was noted except for moderate expression on perinuclear structures (75, 176). The later distribution is consistent with a recent report of IP3-induced Ca2+ release from isolated nuclei prepared from mouse pancreatic acinar cells (47). Studies where IP3 was released from a caged precursor in various localized regions of mouse acinar cells has also functionally confirmed that the apical region of the cell is more sensitive to IP3 than the basal area of the cell (38). These data were later confirmed by a study imaging permeabilized pancreatic acini (70). Quantitative Western analysis and PCR have indicated that there is approximately equal expression of IP3R3 and IP3R2 in pancreatic acinar cells making up ~ 90% of the total complement of IP3R (28, 166). The pivotal importance of IP3R2/3 for Ca2+ signaling and subsequent exocrine secretion has been confirmed by studies of transgenic animals in which both subtypes have been knocked out (42). Individual knock down of IP3R2 or IP3R3 resulted in a modest reduction in the magnitude of Ca2+ release (42, 103) but no obvious global phenotype. However, the compound IP3R2/3 null animal dies soon after weaning. The lack of viability results from a failure to ingest and subsequently digest food as a direct consequence of a general failure of exocrine gland secretion. In both salivary and pancreatic acinar cells from IP3R2/3 null animals, secretagogue-stimulated Ca2+ signals and secretion of fluid and protein are essentially absent (42). These data also reveal that the residual IP3R-1 in the IP3R-2/3 null animal cannot compensate for the loss of the other subtypes in pancreatic acinar cells.
Figure 4. Localization of IP3R in pancreatic slices. The localization of IP3R1 (A) IP3R2 (B) and IP3R3 (C) was determined with specific antibodies to individual InsP3R types and visualization by confocal microscopy. IP3R of all types predominately localized to the extreme apical pole of acinar cells, immediately below the luminal plasma membrane (arrows in A-D; compare localization of zymogen granules visualized by staining for amylase in panel D). IP3R1 and IP3R3 also localized to perinuclear structures (arrowheads in A and C). (From reference (176)). (E) IP3R3 (stained in red) are colocalized with the terminal web of the actin cytoskeleton (stained in green).
IP3R Structure and Regulation
The functional IP3R is formed co-translationally by the tetrameric association of four individual receptor subunits (112, 140). In pancreatic acinar cells there is evidence that the channel can form a heterotetramer since multiple types of IP3R can be detected in immunoprecipitates of specific individual receptor types (1, 23, 167). Indeed, a recent study has shown by sequential immunodepletion of individual subtypes from pancreatic lysates, that homotetrameric IP3R likely constitute a minority of the functional channels (1). Each subunit has a binding site for InsP3 towards the n-terminus which is formed by a cluster of positively charged amino acids thought to coordinate the negatively charged phosphate groups of IP3 (11, 125). The Kd for binding of IP3 to pancreatic membranes has been reported to be between 1-7 nM, a figure similar to that reported for other peripheral tissues such as liver (39, 50). The c-terminus of each subunit is postulated to span intracellular membranes six times and a single cation selective pore is formed from this region of the protein in the tetrameric receptor. When purified or expressed in a heterologous system and then reconstituted in planar lipid bilayers the protein can be demonstrated to function as an InsP3 gated cation channel with many of the characteristics of the release channel (34). The fact that the outer nuclear membrane is continuous with the ER has been exploited in patch-clamp experiments to study IP3R channel activity in isolated nuclei from Xenopus laevis oocytes, COS and DT40-3KO cells (10, 87, 159). These experiments have provided insight into the activity and regulation of the channel in a native membrane, however to date, no information is available regarding the activity of IP3R in native pancreatic membranes.
While the IP3 binding pocket and channel pore are highly conserved between IP3R family members, the intervening sequence between the binding region and pore is more divergent and consists of the so-called “regulatory and coupling” or “modulatory” domain. This region consisting of ~1600 amino acids is thought to be important in modulating the Ca2+ release properties of the IP3R. Indeed, Ca2+ release through the IP3R is markedly influenced by many factors, most importantly by Ca2+ itself (9, 36). The majority of studies have indicated that all forms of the IP3R are biphasically regulated by Ca2+. [Ca2+] in the range 0f 0.5- 1 μM increases the steady-state open-probability of the channel while at higher concentrations the activity decreases (9, 36). This property of the IP3R is thought to be fundamentally important in the generation of the different spatial and temporal pattern of Ca2+ signals observed in cells (141). In pancreatic acinar cells IP3-induced Ca2+ release has been shown to be inhibited when Ca2+ is elevated and enhanced when Ca2+ is buffered with chelators (33, 187). The [Ca2+]i, together with the range of action of Ca2+ can be manipulated by dialyzing cells with buffers exhibiting differing on-rates for Ca2+ binding. In pancreatic acinar cells, restriction of the range of action of Ca2+ using the slow on-rate buffer EGTA resulted in spatially restricted IP3-induced spikes and the attenuation of global waves consistent with EGTA inhibiting the positive effect of Ca2+ to facilitate Ca2+ release between spatially separated release sites. In contrast, the fast on-rate buffer BAPTA resulted in larger monotonic Ca2+ release and this was interpreted to reflect the loss of local Ca2+ inhibition of IP3R (67).
IP3R activity is also influenced through interaction with numerous factors such as proteins, adenine nucleotides and phosphorylation in particular by cyclic nucleotide dependent kinases (for review see (112, 113). The IP3R1 represents one of the major substrates for phosphorylation by protein kinase A in brain and thus represents a potentially important locus for cross-talk between the cAMP and Ca2+ signaling systems (116). In pancreatic acinar cells, PKA activation results in phosphorylation of IP3R3 (72, 133). Functionally, phosphorylation of IP3R in pancreatic acinar cells correlates with IP3-induced Ca2+ release which is decreased in terms of the magnitude and kinetics of Ca2+ release (51, 133).
Physiologically relevant concentrations of CCK, but not ACh also result in PKA-dependent phosphorylation of IP3R (133). This observation is consistent with earlier reports that CCK stimulation leads to an increase in cAMP and PKA activation. CCK-induced phosphorylation of InsP3R may contribute to the specific characteristics of CCK-induced Ca2+ signals as maneuvers which interfere with PKA activation disrupt the pattern of CCK-induced but not ACh–mediated Ca2+ signaling. Conversely, raising cAMP converts ACh-induced Ca2+ signaling characteristics into signals which resemble CCK stimulation (16, 51). CCK-stimulated signaling is not affected by raising cAMP or by stimulation with VIP presumably because PKA activation and phosphorylation of IP3R has already occurred (51, 153). CCK and bombesin stimulation result in Ca2+ signals with similar characteristics and this may be related to the fact that bombesin stimulation also results in phosphorylation of IP3R in mouse pancreatic acinar cells (90, 133). Paradoxically, although there are numerous examples of similar attenuated Ca2+ signaling following PKA phosphorylation in other cell types (175), PKA phosphorylation of individual IP3R subtypes studied in isolation has only been shown to increase Ca2+ release (175). These data raise the possibility that the physiologically relevant phosphorylation event actually occurs on a tightly associated binding partner to inhibit Ca2+ release and not the IP3R-3 directly. In vitro, IP3R can also be phosphorylated by protein kinase C, Ca2+/calmodulin dependent kinase II and tyrosine kinases of the src family (112, 113). While no direct evidence has been reported regarding phosphorylation of IP3R by these pathways in pancreatic acinar cells activation of PKC has been shown to inhibit Ca2+ release in permeabilized pancreatic acinar cells and to attenuate Ca2+ oscillations stimulated by secretagogues or by direct G-protein activation, without an effect on PI hydrolysis (165, 181). Thus, the possibility exists that the IP3R is a substrate for other kinases in pancreatic acinar cells.
Cellular levels of ATP also modulate IP3- induced Ca2+ release in permeabilized mouse pancreatic acinar cells (108). ATP has characteristic distinguishing effects on individual IP3R subtypes (175) and despite an approximately equal complement of IP3R2 and IP3R3 in acinar cells, the modulation has essentially identical properties to IP3R2 in wild type animals and is indistinguishable from IP3R3 in IP3R2 null animals. These data indicate that the specific properties of the IP3R2 appear to dominate the overall profile of IP3-induced release in pancreatic acinar cells (108) and are consistent with the effects of ATP occurring on heterotetrameric IP3R in which IP3R2 is present (1). This hypothesis was directly tested in a heterologous expression system where concatenated IP3R constructs consisting of tandem cDNAs encoding defined heterotetramers were expressed in IP3R null cells. The characteristics of Ca2+ release from heterotetrameric IP3R consisting of at least 2 subunits of IP3R2 were indistinguishable from homotetrameric IP3R2 (1, 22).
Introduction of G-protein βγ subunits into pancreatic acinar cells has also been shown to induce Ca2+ release. An initial report attributed this to the activation of PLC β2 by βγ and the subsequent production of IP3 (170). A later report however demonstrated that the Gβγ-induced Ca2+ release was independent of InsP3 production and the result of a direct interaction of Gβγ with IP3R (183). This association was confirmed by co-immunoprecipitation and shown to increase the open-probability of IP3R in a manner independent of IP3. This interaction of IP3R and βγ may be important for the action of Gαi linked agonists in pancreatic acinar cells.
Ryanodine Receptor-Induced Ca2+ Release
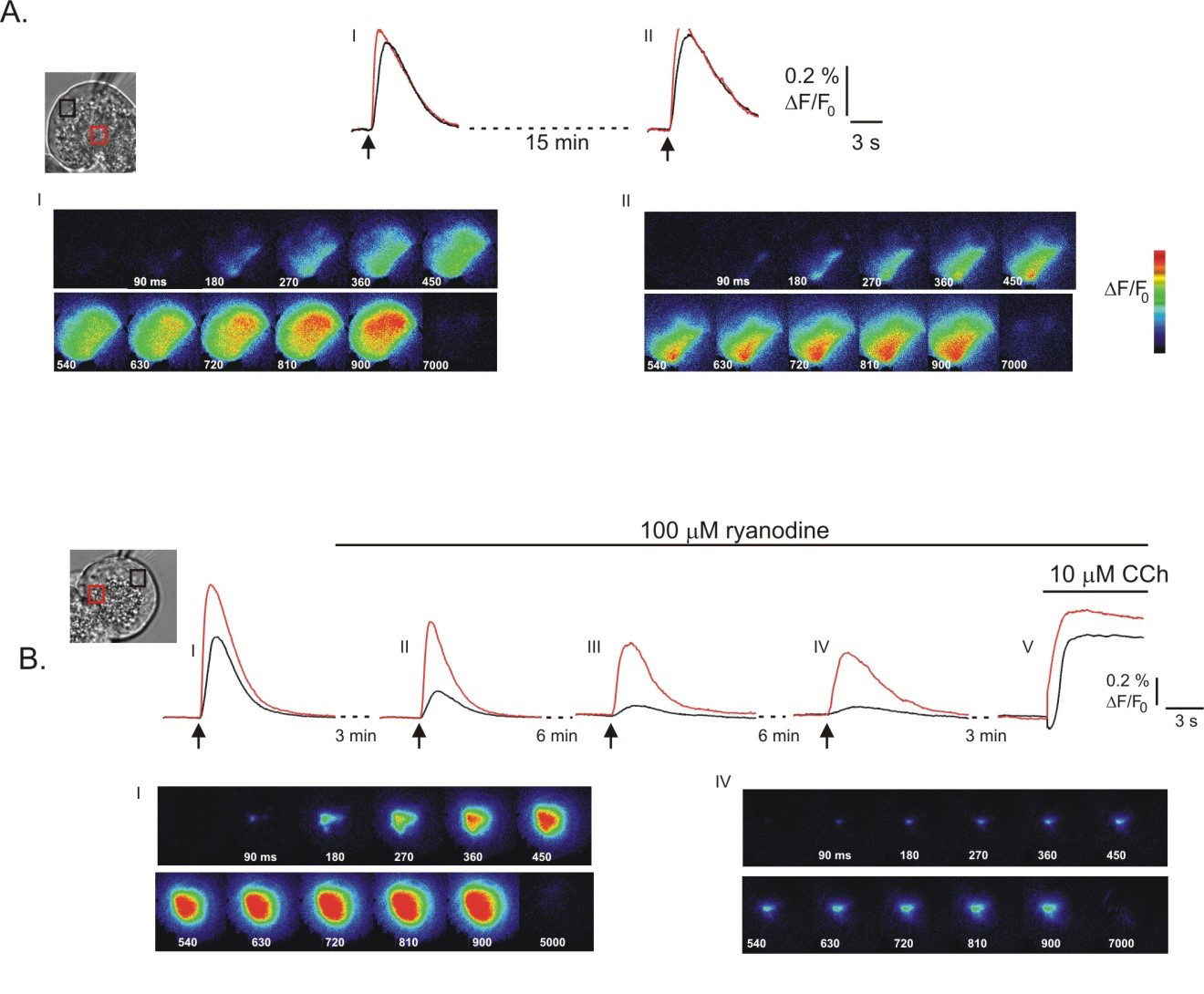
Evidence exists for Ca2+ release initiated through activation of ryanodine receptors (RyR) in pancreatic acinar cells. This family of channels, best studied as the Ca2+ release channel in skeletal and cardiac muscle is classified as belonging to the same gene super-family as IP3R. Indeed, RyR are modulated by similar regulators and share some sequence homology with the IP3R especially in the putative Ca2+ conducting pore region of the c-terminus. However, while IP3R have an absolute requirement for IP3 with Ca2+ as an important co-agonist for gating, RyR only require Ca2+ to open through a process termed calcium-induced calcium release (CICR). The functional expression of RyR in pancreatic acinar cells has been indicated by a number of studies; for example microinjection of Ca2+ in the presence of the IP3R antagonist heparin results in Ca2+ release in mouse pancreatic acinar cells (66). In addition, treatment of pancreatic acinar cells with high concentrations of ryanodine known to block RyR dampens secretagogue-induced Ca2+ signals (101, 134) and one report has shown that low concentrations of ryanodine, which permanently opens the RyR in a sub-conductance state results in Ca2+ release (4). In muscle cells, caffeine activates RyR and results in emptying of sarcoplasmic reticulum Ca2+ stores. In contrast, the majority of reports from pancreatic acinar cells indicate that caffeine does not elicit Ca2+ release and actually inhibits secretagogue induced Ca2+ signaling through an action to inhibit phospholipase C and IP3R-mediated Ca2+ release (4, 151). This later effect of caffeine, together with presumably much lower numbers of RyR in pancreatic acinar cells probably explains the absence of caffeine-induced Ca2+ release.
The physical presence of RyR, has however, been difficult to demonstrate in pancreatic acinar cells with conflicting positive and negative reports of expression. For example, in one study RyR could be detected using western analysis in rodent salivary gland acinar cells but not in pancreatic acinar cells (74). In contrast, an initial study reported the expression using PCR analysis of RyR type 2 (RyR2) but not RyR type 1 or 3 (RyR1 and RyR3 respectively) in mRNA extracted from rat pancreatic acini (77). A later report however, using single cell PCR and western analysis demonstrated that all three types of RyR were expressed in pancreatic acini (37). A study performed in human acini has reported mRNA for RyR1, but not RyR2/3. Notably, [3H]-ryanodine binding to membrane preparations provides evidence that the RyR protein is indeed expressed in human pancreatic acini (78). In all probability, the lack of consensus in these studies relates to the relatively low expression of RyR in pancreas when compared to muscle cells. While IP3R have a well-defined localization in pancreatic acinar cells, several studies have reported that ryanodine receptors have a more diffuse distribution. Immunohistochemistry and studies with fluorescently labeled ryanodine have indicated that RyR are distributed throughout acinar cells with perhaps the greatest concentration in the basal aspect of the cell (61, 77, 134). As a result of this localization to areas of the cell with low levels of IP3R it has been suggested that activation of RyR plays an important role in the propagation of Ca2+ signals from the initial release of Ca2+ in the trigger zone to the basal aspects of the cell. In support of this contention, high concentrations of ryanodine in some studies have been shown to slow or spatially limit the spread of Ca2+ waves in mouse pancreatic acinar cells (Figure 5A and B) (101, 134) and also to selectively attenuate the peak [Ca2+] in the basal aspects of cells (61).
Figure 5. Contribution of RyR to global Ca2+ signals in pancreatic acinar cells. Global Ca2+ signals were initiated by photolysis of caged-IP3. In (A) the images and kinetic traces show that global Ca2+ signals can be initiated multiple times following exposure to 1,4,5-IP3. However, as shown in (B) exposure to a high concentration of ryanodine, known to inhibit RyR leads to a slowing in the progression of the Ca2+ wave, and to the restriction of the signal to the apical pole of the cell following stimulation with 1,4,5-IP3 (compare (B) I; absence of ryanodine and (B) IV; exposure to ryanodine for 15 mins). (From reference (134)).
Ca2+ Release Stimulated by cADPR
In a number of cell types there is evidence that in addition to activation by Ca2+ the RyR activity are modulated by cyclic adenosine diphosphoribose (cADPr). cADPr was first suggested to be a Ca2+ releasing second messenger based on experiments performed in sea-urchin eggs where it was shown to release Ca2+ and function as a messenger during fertilization (26). Subsequently, it has been shown to release Ca2+ and satisfy some of the criteria for a second messenger in mammalian systems including lymphocytes, pancreatic β cells and cardiac myocytes (27, 55, 139). Ca2+ release following cADPr exposure is inhibited by blocking concentrations of ryanodine and appears to reduce the threshold for CICR through RyR (73). Opinion is however divided as to whether it functions through a direct effect on the RyR, or indirectly through interaction with an accessory protein such as FKBP 12.6 or calmodulin (8, 128).
In pancreatic acinar cells the intracellular application of cADPr results in Ca2+ release. This observation has been reported in both rat and mouse acini with either whole cell pipette dialysis of cADPr, liberation of cADPr from a caged precursor by 2 photon flash photolysis and by direct application to permeabilized acini (18, 20, 70, 76, 147). In addition cADPr has been reported to release Ca2+ from a rat pancreatic microsomal preparation (106). The spatial localization of cADPr-induced Ca2+ release remains to be conclusively resolved. In an initial report, cADPR introduced by dialysis from a patch-clamp pipette into mouse pancreatic acinar cells resulted in Ca2+ release from the apical pole of the cell (147). This Ca2+ release was blocked by both ryanodine and interestingly the IP3R antagonist heparin. These data were interpreted as indicating that Ca2+ release mediated by cADPr was dependent on both IP3R and RyR, presumably as highly localized Ca2+ release initially through RyR sensitized neighboring IP3R to basal levels of IP3 (147). Consistent with this view it was also shown that low concentrations of ryanodine also resulted in Ca2+ release from the apical pole in a manner apparently dependent on functional IP3R (4). In contrast, in a study more consistent with the localization of the majority of RyR, selective local uncaging of cADPr in different regions of an acinar cell by 2-photon photolysis reported that the basal aspect of the cell was more sensitive to cADPr (76). These data are consistent with a report that in permeabilized acini the apical pole exhibited a higher affinity for IP3 but cADPr released Ca2+ exclusively from the basal aspects of the cell (70).
Important questions to address which are required to establish cADPr as a bona fide Ca2+ releasing second-messenger in pancreatic acinar cells are to demonstrate that the molecule is produced following secretagogues stimulation and that it is necessary for Ca2+ release. To these ends it has been reported that CCK and ACh but not bombesin stimulate the activity of a cytosolic ADP-ribosyl cyclase activity resulting in the production of cADPr (132). One such enzyme which possesses this activity is CD38. Ca2+ signaling events in acinar cells prepared from CD38 null mice are dampened and appear reminiscent of RyR blockade (40). In addition, 8-NH2-cADPr, a structural analog of cADPr which antagonizes the effect of cADPr has been reported to block CCK and bombesin but not ACh or IP3 –induced Ca2+ signals in mouse pancreatic acinar cells (13, 19). This later result, while not internally consistent with the ability of ACh to induce the formation of cADPr has been suggested to indicate that CCK stimulation preferentially couples to the generation of cADPr and thus may account for some of the distinct characteristics of CCK-induced Ca2+ signals reported.
Nicotinic Acid Dinucleotide Phosphate-Induced Ca2+ Release
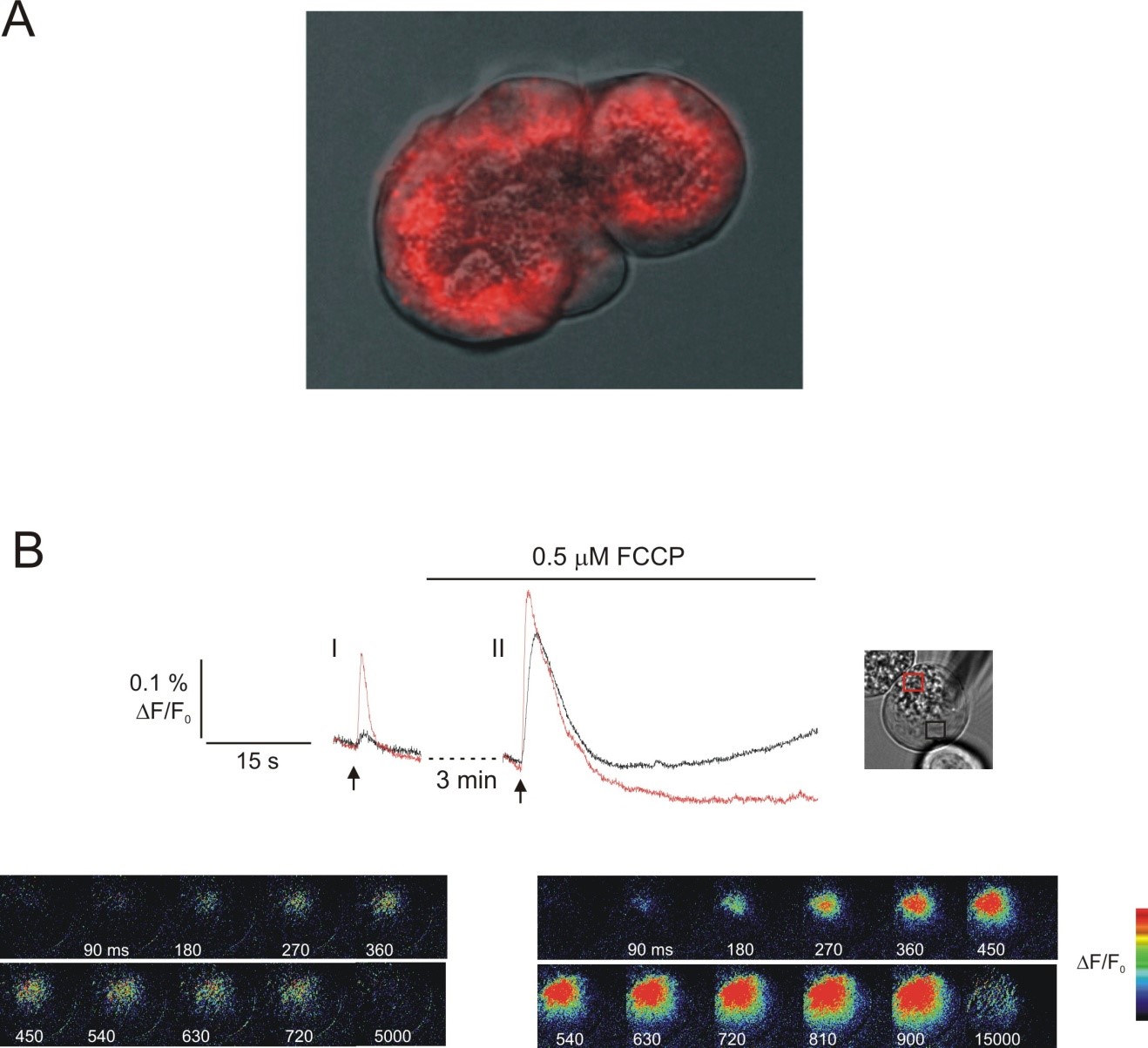
An additional putative messenger that potently induces Ca2+ release in pancreatic acinar cells is nicotinic acid adenine dinucleotide phosphate (NAADP). Once again the activity of this agent was first reported to play a role in invertebrate fertilization. Although it has been reported that NAADP-induced Ca2+ release from nuclei isolated from mouse pancreatic acini is dependent on RyR (47) the majority of evidence suggests that the receptor is likely to represent a novel Ca2+ release channel since while the activity of IP3R and RyR exhibits a bell-shaped dependence on Ca2+ the putative NAADP receptor does not support CICR (43). While this property of NAADP is not well suited to play a role in the propagation of Ca2+ waves it has been suggested that NAADP is required to initiate Ca2+ signals and this initial Ca2+ increase subsequently recruits IP3R and RyR. This idea is supported by the observation that NAADP introduced via the patch pipette into mouse pancreatic acinar cells results in Ca2+ release in the apical pole but that this release is absolutely dependent on both InsP3R and RyR (18). These data suggest that Ca2+ release through NAADP is quantitatively very small but ideally localized to sensitize IP3R and RyR. While IP3R and RyR reside in the ER it has been suggested that NAADP primarily acts on a distinct store probably an acidic lysosome related organelle (172). This idea is primarily based on the observation that NAADP-induced Ca2+ signaling but not cADPr or IP3R-induced Ca2+ release is inhibited by experimental maneuvers which either inhibit vacuolar type H+ ATPase or result in osmotic disruption of lysosomes (172).
The concentration-response relationship for NAADP-induced Ca2+ release also displays unique properties. NAADP-induced Ca2+ release is biphasic; nanomolar levels induce Ca2+ release while micromolar concentrations fail to release Ca2+ but render the mechanism refractory to subsequent stimulation (17). Provocatively, exposure of mouse pancreatic acinar cells to inactivating concentrations of NAADP also renders cells refractory to stimulation with threshold concentrations of CCK but not ACh or bombesin and disruption of the NAADP releasable store selectively disrupts CCK-induced Ca2+ signals (13, 17). In addition, NED-19 an inhibitor of NAADP action attenuates CCK-, but not ACh- induced Ca2+ responses (44). These data suggest that CCK and NAADP under these conditions may utilize a common mechanism to induce Ca2+ release.
In support of this contention, physiologically relevant concentrations of CCK can also be shown to result in the production of NAADP while ACh stimulation does not result in measurable accumulation (173). The molecular mechanism responsible for coupling CCK- receptor occupation to NAADP production is at present not completely understood. Evidence suggests however, that the enzyme CD38 may also be a candidate for the hormone-responsive NAADP synthase because CCK stimulated NAADP formation is absent in CD38 null transgenic animals (40). This activity of CD38 occurs at acidic pH and this requirement appears to be fulfilled by the expression of some of the enzyme in an endosomal compartment (40).
The Two Pore Channel (TPC) family of proteins has recently been proposed as a candidate for the cognate receptor for NAADP (14, 43). The TPC gene family is related to the super-family of voltage gated ion channels and consists of three distinct gene products which are represented in both plants and animals (14). The primary supporting evidence for the idea that TPC represent a NAADP receptor is that the expression of TPCs appears to confer Ca2+ release activity in the presence of NAADP (14, 43, 124). Additionally, the expression of TPCs also leads to specific binding activity of radio-labeled NAADP and TPCs are primarily expressed in an endo-lysosomal compartment, consistent with the prominent site of NAADP-induced Ca2+ release (14, 43). Since this specific binding is much lower affinity than the functional effects of NAADP and specific binding can be observed to co-immunoprecipitated proteins (80, 163), it is questionable whether the effects of NAADP are mediated through an accessory protein. In support of a role for TPC2 as a target of NAADP- Ca2+ release was reduced in acini prepared from TPC-2 knockout animals (44). Of note NAADP-induced Ca2+ release was also reduced in RYR3 knockout animals and in permeabilized cells incubated with α-RYR1 antibodies. The latter observation is consistent with reports from T lymphocytes that suggest RYR are the target of NAADP (31). In total, these data led the authors to suggest that both RYRs and TPC channels might function in pancreatic acinar cells to provide the trigger for Ca2+ release following CCK stimulation.
B. Cellular Mechanisms Underlying Ca2+ influx Across the Plasma Membrane
Ca2+ influx from the extracellular space is essential to sustain Ca2+ signaling in pancreatic acinar cells and in turn the long term maintenance of secretion (152). This influx is not blocked by antagonists of voltage-gated Ca2+ channels, but is attenuated by Lanthanides (107, 152). Ca2+ influx is also sensitive to changes in extracellular pH, being enhanced by alkaline and inhibited in acidic conditions (107, 152).
Store-Operated Ca2+ Entry and its Molecular Components
Functionally, Ca2+ influx can be initiated by substantial depletion of intracellular Ca2+ pools, the so called “store-operated” or “capacitative” Ca2+ entry (SOCE) pathway (119). This pathway can be readily demonstrated by inhibition of ER Ca2+ pumps with the plant sesquiterpene lactone thapsigargin, which results in Ca2+ influx independent of receptor activation and PI hydrolysis (92, 119, 152, 178). Recently, significant progress has been made regarding the molecular candidates responsible for the SOCE pathway. Using siRNA screens the first breakthrough was the identification of stromal interaction molecule 1 (STIM-1) as the ER Ca2+ sensor responsible for coupling intracellular Ca2+ store depletion to the opening of the Ca2+ permeable conductance in the plasma membrane (81, 122, 188). STIM-1 is an integral ER membrane protein, which harbors an EF hand type Ca2+ binding domain localized in the ER lumen. Following depletion of ER stores, Ca2+ dissociates from the EF hand and this results in the aggregation of STIM-1 molecules to areas of the ER close to the plasma membrane where it can physically gate the ion channel responsible for mediating Ca2+ influx (84, 169). Strong candidates for the actual pore forming components of the pathway have also been identified. Using siRNA screens and complimented by linkage analysis of patients with severe combined immunodeficiency, studies have identified members of the Orai family of proteins as the channel constituents of the archetypal SOCE current, the so-called, calcium release activated current (ICRAC). The channel has been extensively characterized in lymphocytes as a highly selective Ca2+ channel (35, 59, 154). In particular, homomultimers of Orai1 form channels with the biophysical characteristics of ICRAC (57). Both STIM-1 and Orai-1 are expressed in mouse pancreatic acinar cells and thus represent good candidates for mediating SOCE (56, 59, 86). Lower levels of secretagogue stimulation result in only modest ER Ca2+ depletion which may not be sufficient to activate STIM-1. While not directly demonstrated in pancreatic acinar cells, minor store depletion may be sensed by STIM-2, a family member which because of increased affinity for ER [Ca2+ ] can activate Orai channels following physiological stimulation (12).It is, however, clear that marked global depletion of ER Ca2+ occurs upon stimulation with maximal concentrations of secretagogues and upon exposure to pancreatic toxins, such as bile acids and fatty acid esters of ethanol (45, 46, 111). Thus, the SOCE pathway may be important during strong stimulation and during pathological Ca2+ depletion of ER stores. Indeed, it has been suggested that activation of this pathway is responsible for the inappropriate intracellular activation of trypsin which occurs in models of acute pancreatitis (71, 97, 98, 120). Consistent with this idea, inhibition of Ca2+ influx with pharmacology that targets SOCE has been shown to reduce the severity of pancreatitis in several experimental animal disease models (48, 162, 164) as well as mitigating the detrimental cellular hallmarks of pancreatitis ex vivo (155). Further, maneuvers which inhibit Ca2+ release, including blockade of IP3R with caffeine which prevent depletion of the ER and thus activation of SOCE, also are protective in experimental pancreatitis (60).
Contribution of TRP Channels
In exocrine cells there is also evidence that members of the canonical Transient Receptor Potential channel family (TRPC) contribute to Ca2+ influx following store depletion (59, 68, 82). This channel, while Ca2+ permeable is a non-selective cation channel, exhibiting significant Na+ permeability. STIM-1 has been reported to also function as the ER calcium sensor for this class of proteins (168). Further, there is molecular and biochemical evidence to suggest that a tertiary complex of STIM-1/TRPC/Orai-1 may contribute to the native SOCE in acinar cells (59, 102). TRPC 3 and 6 are expressed in pancreatic acinar cells (68). In TRPC 3 null transgenic animals, the magnitude of secretagogue- stimulated Ca2+ influx is reduced and this is associated with reduced agonist stimulated amylase secretion (68). In addition, the frequency of agonist-stimulated Ca2+ oscillations was reduced in TRPC 3 null animals supporting the view that Ca2+ influx through this channel may also serve to maintain Ca2+ oscillations (68). Interestingly the absence of TRPC 3 is also protective in animal models of pancreatitis indicating that SOCE may be deleterious under these conditions (68).
While the spatial properties of the Ca2+ signal can be largely attributed to the localization of the Ca2+ release machinery, recent data has also suggested that the distribution of the components of the SOCE Ca2+ influx pathway may also be spatially heterogeneous. The majority of Orai-1 and TRPC3 is reported to be localized to the apical and lateral plasma membrane (56, 58, 68, 86). One study has shown that following store depletion some STIM-1 translocates to the apical domain and is colocalized with Orai-1. Further, this study demonstrated that Ca2+ influx following store depletion could be detected earlier in the apical region when compared to more basal aspects of the cell (59). Of note, a majority of STIM-1 does not co-localize with Orai-1 following store depletion (59, 86). Indeed, a study monitoring the localization of adenovirally expressed fluorescently tagged STIM-1 reported little co-localization with Orai-1 and that the vast majority of STIM-1 translocated to the basolateral aspects of cells following stimulation (86). Further work will be necessary to unequivocally confirm the cellular localization of this influx pathway in acinar cells.
Contribution of ARC Channels
As stated earlier, during secretagogue stimulation only relatively minor depletion of the ER pool as a whole occurs (111). This observation raises the question whether SOCE operates under physiological conditions. If global ER Ca2+ is not markedly reduced during physiological stimulation, it follows that for SOCE to be activated under these conditions, local depletion of the strands of ER that infiltrate the apical domain (49) which are replete with IP3R (176) must occur allowing the activation of STIM1/2. A contrary view is that a different mechanism largely controls Ca2+ influx during physiological stimulation. In support of this idea, an electrophysiological study of mouse pancreatic acinar cells failed to detect SOCE currents following stimulation with physiologically relevant concentrations of agonists. Under these conditions it was reported that a channel activated by arachidonic acid was predominately responsible for Ca2+ influx (93). This conductance has been termed IARC and is present in many non-excitable cell types (127, 145). Notably, this channel also requires STIM-1 for activation and the pore is formed from multimers of Orai1 and Orai3 (146).
C. Cellular Mechanisms Underlying Ca2+ Clearance from the Cytosol
Following [Ca2+]i elevation, mechanisms must be present to reduce [Ca2+]i rapidly and efficiently during the falling phase of Ca2+ oscillations and ultimately to terminate Ca2+ signals following secretagogue removal. Moreover, the resting [Ca2+]i of 50-200 nM must be maintained in the face of a large concentration gradient from the cell exterior, leak from the ER and the negative intracellular potential, all of which would tend to drive Ca2+ into the cytoplasm. Homeostasis is accomplished by a variety of pumps and transporters that have specific distribution on both the plasma membrane and on the membranes of various organelles.
Ca2+ Pumping Across the PM
Tepikin and colleagues employing a technique where the extracellular [Ca2+] is monitored by an indicator in a small volume of extracellular fluid demonstrated that Ca2+ is extruded across the plasma membrane following agonist stimulation (142, 144). This in all probability occurs by Ca2+ pumps of the plasma membrane Ca2+-ATPase gene family (PMCA). Ca2+-ATPase activity is present in plasma membrane preparations isolated form pancreatic acinar cells and immunoblotting demonstrates the expression of PMCA family members (2, 6, 74). Upon supermaximal stimulation with CCK or ACh the amount of Ca2+ lost from the cell approximates the entire agonist-releasable pool (143). Ca2+ extrusion also occurs during more physiological stimulation with CCK and the activity follows the [Ca2+]I (144). PMCA is also important for maintaining resting [Ca2+]i since it is activity can be demonstrated at basal [Ca2+]i. Indeed the rate of pumping has been shown to have a steep dependence on [Ca2+]I (Hill coefficient of ~3) and is effectively saturated at [Ca2+]above 400 nM (15). The activity of the PMCA can also be increased by agonist stimulation in a manner independent of Ca2+ and this may reflect modulation of the pumps activity by phosphorylation or association with regulatory proteins (186). PMCA is not homogeneously distributed over the entire plasma membrane and appears to be most abundant in the luminal and lateral plasma membrane (74). The localization of PMCA correlates with the site of most apparent Ca2+ pumping activity since Ca2+ extrusion occurs preferentially from the apical aspects of mouse pancreatic acinar cells when monitored using an indicator with limited diffusional mobility (7).
Ca2+ Uptake into the ER
Ca2+ pumps are also expressed on ER membranes (117, 160). Indeed, the ER in pancreatic acinar cells has been shown to effectively function as a single continuous Ca2+ store (95, 111). This has been best illustrated by demonstrating that ER Ca2+ pump activity, exclusively at the basal pole can recharge the ER following maximal agonist stimulation such that Ca2+ signals can be initiated at the apical pole (95). ER Ca2+-ATPases belong to the sarcoplasmic and endoplasmic reticulum Ca2+ ATPase gene family (SERCA). Both SERCA 2A and SERCA 2B have been reported to be expressed in pancreatic acinar cells and specific sub-cellular distributions of specific SERCA pumps have been reported (74). SERCA 2A appears to be distributed very similar to InsP3R exclusively in the apical pole while SERCA 2B predominately resides in the basal aspects of the cell. All SERCA pumps are inhibited by thapsigargin and treatment results in Ca2+ leak from the ER store. In pancreatic acinar cells, thapsigargin treatment results in a uniform rise in [Ca2+]i and abolishes the characteristic secretagogue-stimulated apically initiated Ca2+ wave even before the ER is fully depleted (74). The later observation indicates that the microenvironment created by SERCA pumps is crucial for the initiation of Ca2+ signals in the apical pole during Ca2+ oscillations. This could occur as a function of the SERCA pump controlling either the cytosolic [Ca2+] or local luminal ER [Ca2+] in the apical pole as both SERCA pumps and IP3R are markedly influenced by the [Ca2+]on both faces of the ER membrane (96, 174).
Ca2+ Uptake by Mitochondria
A further Ca2+ uptake mechanism that plays a significant role in spatially shaping Ca2+ signals in pancreatic acinar cells is through Ca2+ sequestration into mitochondria. Mitochondrial Ca2+ uptake occurs as a function of the large electrical potential across the inner mitochondrial membrane via a ruthenium red sensitive Ca2+ uniporter (21). Because the Ca2+ uniporter is a relatively low affinity, high capacity transporter, it was generally believed that mitochondrial Ca2+ uptake was only relevant under pathological conditions when the [Ca2+] was significantly elevated for prolonged periods of time. Recently, however, with the advent of mitochondrially targeted indicators, this idea has been reevaluated given that it has become clear that mitochondria function during normal physiological Ca2+ signaling in many cell types (121). Moreover, it is now thought that mitochondrial Ca2+ uptake is important not only for shaping cytosolic Ca2+ signals but also for stimulating the production of ATP as key enzymes in the TCA cycle are Ca2+ dependent (30). It appears that the privileged localization of mitochondria very close to Ca2+ release sites, where Ca2+ is presumably very high allows the Ca2+ uniporter to function effectively under these conditions.
The long sought after molecular components of the mitochondrial uniporter have recently been elucidated. Initially, extensive comparative proteomics for inner mitochondrial membrane proteins present in vertebrates, kinetoplastids (protists containing flagella) but not yeast (which do not have a uniporter) identified MICU1 as essential for mitochondrial uptake (114). Second, with the knowledge that the uniporter complex contained a highly Ca2+ selective channel (69), two groups identified an integral membrane protein, MCU which had co-evolved with MICU1 (5, 29). Together these proteins form the core of a complex, which reconstitutes ruthenium-red sensitive mitochondrial Ca2+ uptake. A recent study utilizing cells prepared from MCU knockout animals has clearly demonstrated that mitochondrial Ca2+ uptake is disrupted, thus providing strong evidence for its role in acini (25). Interestingly, disruption of mitochondrial Ca2+ signaling did not alter the severity of experimental pancreatitis despite compromising mitochondrial bioenergetics.
In pancreatic acinar cells, mitochondria are indeed in close proximity to Ca2+ release sites since stimulation with physiological concentrations of both CCK and ACh which raise global [Ca2+]i only to sub-micromolar levels lead to mitochondrial Ca2+ uptake (54, 109, 156). Furthermore, mitochondrial Ca2+ uptake in pancreatic acinar cells is coupled to the conversion of NAD to NADH (156). Further, stimulation of pancreatic acinar cells with Ca2+ mobilizing secretagogues has been shown to result in the net generation of ATP as reported by adenovirally expressed mitochondrially targeted luciferase constructs (158). In common with other Ca2+ clearance mechanisms in acini, energized mitochondria also have specific sub-cellular localization (Figure 6A) (157). The majority of studies have reported that mitochondria are concentrated in a perigranular “belt” (134, 149), together with additional further sub-populations surrounding the nucleus and immediately below the basolateral plasma membrane (109). Strikingly, It appears that these perigranular mitochondria play a role in limiting the spread of Ca2+ signals upon stimulation with threshold concentrations of agonist as disruption of mitochondrial Ca2+ uptake facilitates the spread of normally spatially confined Ca2+ transients (Figure 6B) (134, 149).
Figure 6. Functional consequences of mitochondrial distribution in pancreatic acini. In (A), the localization of active mitochondria was visualized by confocal microscopy in living mouse pancreatic acini loaded with mitotracker red. (A) shows mitotracker red fluorescence merged with a phase image of a small mouse acini. The predominant localization of mitochondria is to a peri-granular belt surrounding the zymogen granules. (B) photolysis of threshold concentrations of 1,4,5-IP3 result in apically limited Ca2+ signals as shown in the images and kinetic traces in (B) I (red trace apical ROI; black trace basal ROI). In the same cell, following disruption of the mitochondrial membrane potential, and thus mitochondrial Ca2+ uptake, an identical exposure to 1,4,5 IP3 results in a global Ca2+ signal. These data suggest that functional mitochondria are required to constrain Ca2+ signals in the apical portion of the acinar cell. (From reference (134)).
IV. Summary
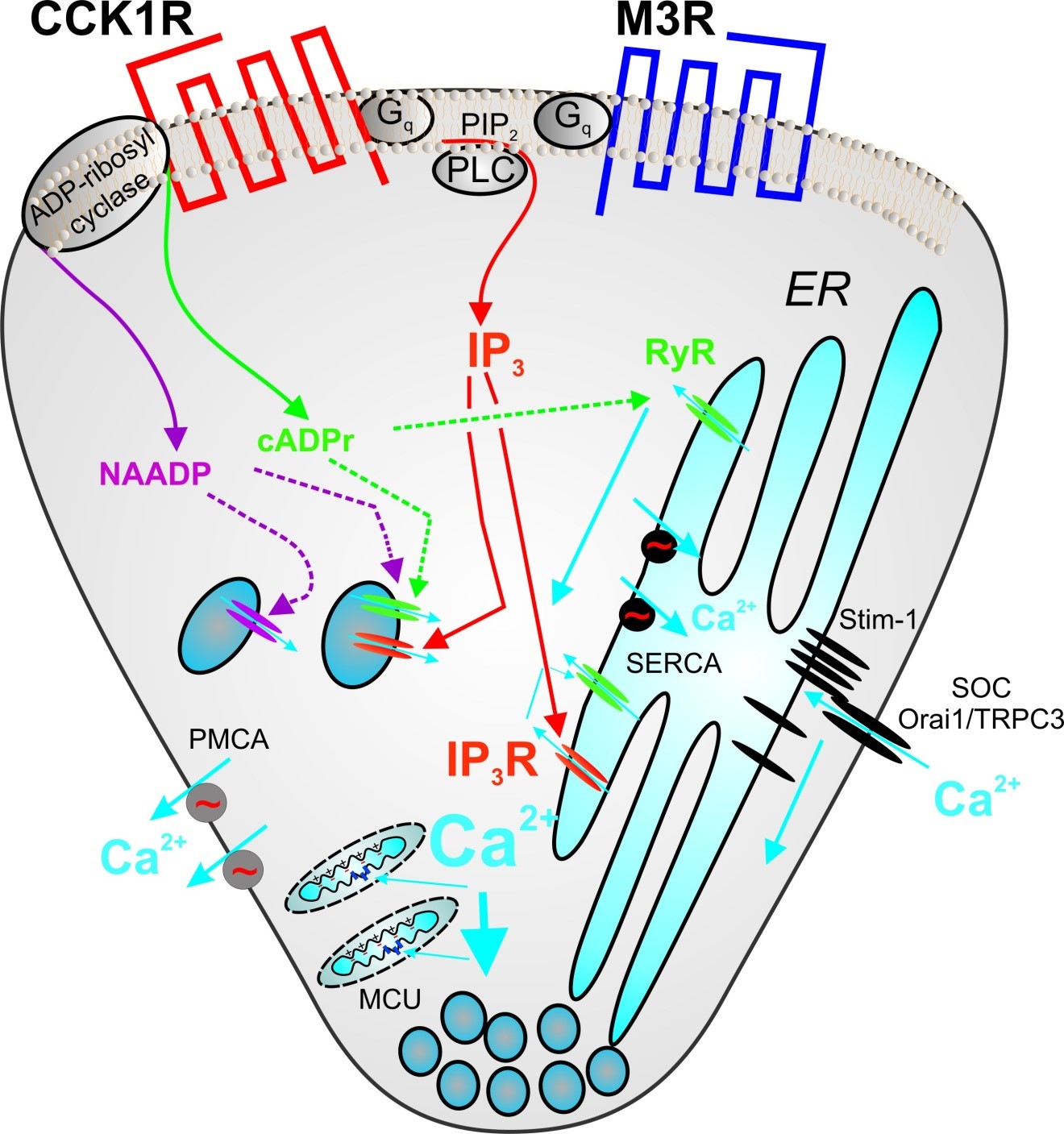
A cartoon summarizing the molecular components currently believed to be involved in the processes of Ca2+ release, Ca2+ influx and Ca2+ clearance from the cytosol of pancreatic acinar cells following secretagogue stimulation is shown in Figure 7.
Figure 7. Intracellular Ca2+ signaling in pancreatic acinar cells is initiated by the binding of Acetylcholine to Muscarinic M3 receptors (M3R) and by Cholecystokinin (CCK) to CCK receptors, predominately the CCK1R in mice and rats. Both receptors are classical seven transmembrane domain receptors coupled to guanine nucleotide-binding proteins (G proteins). Activation of both receptors leads to stimulation of Gαq and increased activity of phospholipase C-β (PLC) which cleaves the membrane phospholipid phosphatidylinositol,4,5,-bisphosphate (PIP2) into diacylglycerol and inositol 1,4,5-trisphosphate (IP3). IP3 diffuses through the cytoplasm and interacts with InsP3 receptors (IP3R), largely type-2 and -3 present predominantly on the apical endoplasmic reticulum (ER) resulting in Ca2+ release into the cytoplasm. Ca2+ release from IP3R acts a co-agonist to increase further IP3R activity and also acts on Ryanodine receptors (RyR) to induce Ca2+ release. Depletion of Ca2+ within the ER results in Ca2+influx from the extracellular space. The ER Ca2+ sensor has been identified as stromal interaction molecule-1 (stim-1). Following ER depletion, Ca2+ is released from an EF hand present in a domain of stim-1 within the ER lumen and this results in aggregation of several stim-1 molecules. Aggregation of stim-1 is sufficient to gate plasma membrane Ca2+ channels and leads to Ca2+ influx. Good candidates for the channel pore are the proteins Orai-1 and TRPC3. CCK receptor stimulation also stimulates ADP-ribosyl cyclase activity resulting in the formation of two additional Ca2+ mobilizing second messengers; Nicotinic acid adenine dinucleotide phosphate (NAADP) and cyclic-ADP ribose (cADPr). The particular cyclase responsible has not been defined in pancreas but good candidates include CD38 and CD157. cADPr is generally thought to act on RyR, while the target of NAADP is currently the subject of intense research. Candidates include the RyR and Two Pore Channel (TPC). In addition to the ER, Ca2+ can also be released from a store, which accumulates Ca2+ in a manner dependent on a proton gradient- known as the “acidic store”. This pool likely represents the endolysosomal compartment. This pool has been reported to be responsive to IP3, cADPr and NAADP and may represent the store initially released following receptor stimulation. Ca2+ is removed from the ccytoplasm by the concerted action of SERCA (ER Ca2+-ATPase), PMCA (PM Ca2+-ATPase) and the action of MCU (mitochondrial uniporter).
V. References
- Alzayady KJ, Wagner LE, 2nd, Chandrasekhar R, Monteagudo A, Godiska R, Tall GG, Joseph SK, and Yule DI. Functional inositol 1,4,5-trisphosphate receptors assembled from concatenated homo- and heteromeric subunits. J Biol Chem 288: 29772-29784, 2013. PMID: 23955339.
- Ansah TA, Molla A, and Katz S. Ca2+-ATPase activity in pancreatic acinar plasma membranes. Regulation by calmodulin and acidic phospholipids. J Biol Chem 259: 13442-13450, 1984. PMID: 6149223.
- Ashby MC, Camello-Almaraz C, Gerasimenko OV, Petersen OH, and Tepikin AV. Long distance communication between muscarinic receptors and Ca2+ release channels revealed by carbachol uncaging in cell-attached patch pipette. J Biol Chem 278: 20860-20864, 2003. PMID: 12657637.
- Ashby MC, Petersen OH, and Tepikin AV. Spatial characterisation of ryanodine-induced calcium release in mouse pancreatic acinar cells. Biochem J 369: 441-445, 2003. PMID: 12444927.
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, and Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476: 341-345, 2011. PMID: 21685886.
- Bayerdorffer E, Eckhardt L, Haase W, and Schulz I. Electrogenic calcium transport in plasma membrane of rat pancreatic acinar cells. J Membr Biol 84: 45-60, 1985. PMID: 3999124.
- Belan PV, Gerasimenko OV, Tepikin AV, and Petersen OH. Localization of Ca2+ extrusion sites in pancreatic acinar cells. J Biol Chem 271: 7615-7619, 1996. PMID: 8631796.
- Berridge MJ, Bootman MD, and Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4: 517-529, 2003. PMID: 12838335.
- Bezprozvanny I, Watras J, and Ehrlich BE. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 351: 751-754, 1991. PMID: 1648178.
- Boehning D, Mak DO, Foskett JK, and Joseph SK. Molecular determinants of ion permeation and selectivity in inositol 1,4,5-trisphosphate receptor Ca2+ channels. J Biol Chem 276: 13509-13512, 2001. PMID: 11278266.
- Bosanac I, Michikawa T, Mikoshiba K, and Ikura M. Structural insights into the regulatory mechanism of IP3 receptor. Biochim Biophys Acta 1742: 89-102, 2004. PMID: 15590059.
- Brandman O, Liou J, Park WS, and Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 131: 1327-1339, 2007. PMID: 18160041.
- Burdakov D, Cancela JM, and Petersen OH. Bombesin-induced cytosolic Ca2+ spiking in pancreatic acinar cells depends on cyclic ADP-ribose and ryanodine receptors. Cell Calcium 29: 211-216, 2001. PMID: 11162858.
- Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, Tang J, Rietdorf K, Teboul L, Chuang KT, Lin P, Xiao R, Wang C, Zhu Y, Lin Y, Wyatt CN, Parrington J, Ma J, Evans AM, Galione A, and Zhu MX. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 459: 596-600, 2009. PMID: 19387438.
- Camello P, Gardner J, Petersen OH, and Tepikin AV. Calcium dependence of calcium extrusion and calcium uptake in mouse pancreatic acinar cells. J Physiol 490 ( Pt 3): 585-593, 1996. PMID: 8683459.
- Camello PJ, Petersen OH, and Toescu EC. Simultaneous presence of cAMP and cGMP exert a co-ordinated inhibitory effect on the agonist-evoked Ca2+ signal in pancreatic acinar cells. Pflugers Arch 432: 775-781, 1996. PMID: 8772126.
- Cancela JM, Churchill GC, and Galione A. Coordination of agonist-induced Ca2+-signalling patterns by NAADP in pancreatic acinar cells. Nature 398: 74-76, 1999. PMID: 10078532.
- Cancela JM, Gerasimenko OV, Gerasimenko JV, Tepikin AV, and Petersen OH. Two different but converging messenger pathways to intracellular Ca(2+) release: the roles of nicotinic acid adenine dinucleotide phosphate, cyclic ADP-ribose and inositol trisphosphate. EMBO J 19: 2549-2557, 2000. PMID: 10835353.
- Cancela JM, and Petersen OH. The cyclic ADP ribose antagonist 8-NH2-cADP-ribose blocks cholecystokinin-evoked cytosolic Ca2+ spiking in pancreatic acinar cells. Pflugers Arch 435: 746-748, 1998. PMID: 9479030.
- Cancela JM, Van Coppenolle F, Galione A, Tepikin AV, and Petersen OH. Transformation of local Ca2+ spikes to global Ca2+ transients: the combinatorial roles of multiple Ca2+ releasing messengers. EMBO J 21: 909-919, 2002. PMID: 11867519.
- Carafoli E. Intracellular calcium homeostasis. Annu Rev Biochem 56: 395-433, 1987. PMID: 3304139.
- Chandrasekhar R, Alzayady KJ, Wagner LE, 2nd, and Yule DI. Unique Regulatory Properties of Heterotetrameric Inositol 1,4,5-Trisphosphate Receptors Revealed by Studying Concatenated Receptor Constructs. J Biol Chem 291: 4846-4860, 2016. PMID: 26755721.
- Chandrasekhar R, Alzayady KJ, and Yule DI. Using concatenated subunits to investigate the functional consequences of heterotetrameric inositol 1,4,5-trisphosphate receptors. Biochem Soc Trans 43: 364-370, 2015. PMID: 26009177.
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, and Kim DS. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499: 295-300, 2013. PMID: 23868258.
- Chvanov M, Voronina S, Zhang X, Telnova S, Chard R, Ouyang Y, Armstrong J, Tanton H, Awais M, Latawiec D, Sutton R, Criddle DN, and Tepikin AV. Knockout of the Mitochondrial Calcium Uniporter Strongly Suppresses Stimulus-Metabolism Coupling in Pancreatic Acinar Cells but Does Not Reduce Severity of Experimental Acute Pancreatitis. Cells 9: 2020. PMID: 32516955.
- Clapper DL, Walseth TF, Dargie PJ, and Lee HC. Pyridine nucleotide metabolites stimulate calcium release from sea urchin egg microsomes desensitized to inositol trisphosphate. J Biol Chem 262: 9561-9568, 1987. PMID: 3496336.
- Cui Y, Galione A, and Terrar DA. Effects of photoreleased cADP-ribose on calcium transients and calcium sparks in myocytes isolated from guinea-pig and rat ventricle. Biochem J 342 ( Pt 2): 269-273, 1999. PMID: 10455010.
- De Smedt H, Missiaen L, Parys JB, Henning RH, Sienaert I, Vanlingen S, Gijsens A, Himpens B, and Casteels R. Isoform diversity of the inositol trisphosphate receptor in cell types of mouse origin. Biochem J 322 ( Pt 2): 575-583, 1997. PMID: 9065779.
- De Stefani D, Raffaello A, Teardo E, Szabo I, and Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476: 336-340, 2011. PMID: 21685888.
- Denton RM, and McCormack JG. Ca2+ as a second messenger within mitochondria of the heart and other tissues. Annu Rev Physiol 52: 451-466, 1990. PMID: 2184763.
- Diercks BP, Werner R, Weidemuller P, Czarniak F, Hernandez L, Lehmann C, Rosche A, Kruger A, Kaufmann U, Vaeth M, Failla AV, Zobiak B, Kandil FI, Schetelig D, Ruthenbeck A, Meier C, Lodygin D, Flugel A, Ren D, Wolf IMA, Feske S, and Guse AH. ORAI1, STIM1/2, and RYR1 shape subsecond Ca(2+) microdomains upon T cell activation. Sci Signal 11: 2018. PMID: 30563862.
- Dormer RL, and Williams JA. Secretagogue-induced changes in subcellular Ca2+ distribution in isolated pancreatic acini. Am J Physiol 240: G130-140, 1981. PMID: 7468805.
- Engling R, Fohr KJ, Kemmer TP, and Gratzl M. Effect of GTP and Ca2+ on inositol 1,4,5-trisphosphate induced Ca2+ release from permeabilized rat exocrine pancreatic acinar cells. Cell Calcium 12: 1-9, 1991. PMID: 2015618.
- Ferris CD, Huganir RL, Supattapone S, and Snyder SH. Purified inositol 1,4,5-trisphosphate receptor mediates calcium flux in reconstituted lipid vesicles. Nature 342: 87-89, 1989. PMID: 2554143.
- Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, and Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441: 179-185, 2006. PMID: 16582901.
- Finch EA, Turner TJ, and Goldin SM. Calcium as a coagonist of inositol 1,4,5-trisphosphate-induced calcium release. Science 252: 443-446, 1991. PMID: 2017683.
- Fitzsimmons TJ, Gukovsky I, McRoberts JA, Rodriguez E, Lai FA, and Pandol SJ. Multiple isoforms of the ryanodine receptor are expressed in rat pancreatic acinar cells. Biochem J 351: 265-271, 2000. PMID: 10998370.
- Fogarty KE, Kidd JF, Tuft DA, and Thorn P. Mechanisms underlying InsP3-evoked global Ca2+ signals in mouse pancreatic acinar cells. J Physiol 526 Pt 3: 515-526, 2000. PMID: 10922004.
- Fohr KJ, Wahl Y, Engling R, Kemmer TP, and Gratzl M. Decavanadate displaces inositol 1,4,5-trisphosphate (IP3) from its receptor and inhibits IP3 induced Ca2+ release in permeabilized pancreatic acinar cells. Cell Calcium 12: 735-742, 1991. PMID: 1663003.
- Fukushi Y, Kato I, Takasawa S, Sasaki T, Ong BH, Sato M, Ohsaga A, Sato K, Shirato K, Okamoto H, and Maruyama Y. Identification of cyclic ADP-ribose-dependent mechanisms in pancreatic muscarinic Ca(2+) signaling using CD38 knockout mice. J Biol Chem 276: 649-655, 2001. PMID: 11001947.
- Furuichi T, Yoshikawa S, Miyawaki A, Wada K, Maeda N, and Mikoshiba K. Primary structure and functional expression of the inositol 1,4,5-trisphosphate-binding protein P400. Nature 342: 32-38, 1989. PMID: 2554142.
- Futatsugi A, Nakamura T, Yamada MK, Ebisui E, Nakamura K, Uchida K, Kitaguchi T, Takahashi-Iwanaga H, Noda T, Aruga J, and Mikoshiba K. IP3 receptor types 2 and 3 mediate exocrine secretion underlying energy metabolism. Science 309: 2232-2234, 2005. PMID: 16195467.
- Galione A, Patel S, and Churchill GC. NAADP-induced calcium release in sea urchin eggs. Biol Cell 92: 197-204, 2000. PMID: 11043408.
- Gerasimenko JV, Charlesworth RM, Sherwood MW, Ferdek PE, Mikoshiba K, Parrington J, Petersen OH, and Gerasimenko OV. Both RyRs and TPCs are required for NAADP-induced intracellular Ca(2)(+) release. Cell Calcium 58: 237-245, 2015. PMID: 26100948.
- Gerasimenko JV, Gerasimenko OV, and Petersen OH. The role of Ca2+ in the pathophysiology of pancreatitis. J Physiol 592: 269-280, 2014. PMDI: 23897234.
- Gerasimenko JV, Gryshchenko O, Ferdek PE, Stapleton E, Hebert TO, Bychkova S, Peng S, Begg M, Gerasimenko OV, and Petersen OH. Ca2+ release-activated Ca2+ channel blockade as a potential tool in antipancreatitis therapy. Proc Natl Acad Sci U S A 110: 13186-13191, 2013. PMID: 23878235.
- Gerasimenko JV, Maruyama Y, Yano K, Dolman NJ, Tepikin AV, Petersen OH, and Gerasimenko OV. NAADP mobilizes Ca2+ from a thapsigargin-sensitive store in the nuclear envelope by activating ryanodine receptors. J Cell Biol 163: 271-282, 2003. PMID: 14568993.
- Gerasimenko JV, Peng S, Tsugorka T, and Gerasimenko OV. Ca(2+) signalling underlying pancreatitis. Cell Calcium 70: 95-101, 2018. PMID: 28552244.
- Gerasimenko OV, Gerasimenko JV, Rizzuto RR, Treiman M, Tepikin AV, and Petersen OH. The distribution of the endoplasmic reticulum in living pancreatic acinar cells. Cell Calcium 32: 261-268, 2002. PMID: 12543088.
- Giovannucci DR, Bruce JI, Straub SV, Arreola J, Sneyd J, Shuttleworth TJ, and Yule DI. Cytosolic Ca(2+) and Ca(2+)-activated Cl(-) current dynamics: insights from two functionally distinct mouse exocrine cells. J Physiol 540: 469-484, 2002. PMID: 11956337.
- Giovannucci DR, Groblewski GE, Sneyd J, and Yule DI. Targeted phosphorylation of inositol 1,4,5-trisphosphate receptors selectively inhibits localized Ca2+ release and shapes oscillatory Ca2+ signals. J Biol Chem 275: 33704-33711, 2000. PMID: 10887192.
- Gonzalez A, Pfeiffer F, Schmid A, and Schulz I. Effect of intracellular pH on acetylcholine-induced Ca2+ waves in mouse pancreatic acinar cells. Am J Physiol 275: C810-817, 1998. PMID: 9730965.
- Gonzalez A, Schmid A, Sternfeld L, Krause E, Salido GM, and Schulz I. Cholecystokinin-evoked Ca(2+) waves in isolated mouse pancreatic acinar cells are modulated by activation of cytosolic phospholipase A(2), phospholipase D, and protein kinase C. Biochem Biophys Res Commun 261: 726-733, 1999. PMID: 10441493.
- Gonzalez A, Schulz I, and Schmid A. Agonist-evoked mitochondrial Ca2+ signals in mouse pancreatic acinar cells. J Biol Chem 275: 38680-38686, 2000. PMID: 10995756.
- Guse AH, da Silva CP, Berg I, Skapenko AL, Weber K, Heyer P, Hohenegger M, Ashamu GA, Schulze-Koops H, Potter BV, and Mayr GW. Regulation of calcium signalling in T lymphocytes by the second messenger cyclic ADP-ribose. Nature 398: 70-73, 1999. PMID: 10078531.
- Gwack Y, Srikanth S, Feske S, Cruz-Guilloty F, Oh-hora M, Neems DS, Hogan PG, and Rao A. Biochemical and functional characterization of Orai proteins. J Biol Chem 282: 16232-16243, 2007. PMID: 17293345.
- Hogan PG, and Rao A. Store-operated calcium entry: Mechanisms and modulation. Biochem Biophys Res Commun 460: 40-49, 2015. PMID: 25998732.
- Hollinger S, and Hepler JR. Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol Rev 54: 527-559, 2002. PMID: 12223533.
- Hong JH, Li Q, Kim MS, Shin DM, Feske S, Birnbaumer L, Cheng KT, Ambudkar IS, and Muallem S. Polarized but differential localization and recruitment of STIM1, Orai1 and TRPC channels in secretory cells. Traffic 12: 232-245, 2011. PMID: 21054717.
- Huang W, Cane MC, Mukherjee R, Szatmary P, Zhang X, Elliott V, Ouyang Y, Chvanov M, Latawiec D, Wen L, Booth DM, Haynes AC, Petersen OH, Tepikin AV, Criddle DN, and Sutton R. Caffeine protects against experimental acute pancreatitis by inhibition of inositol 1,4,5-trisphosphate receptor-mediated Ca2+ release. Gut 66: 301-313, 2017. PMID: 16642860.
- Husain SZ, Prasad P, Grant WM, Kolodecik TR, Nathanson MH, and Gorelick FS. The ryanodine receptor mediates early zymogen activation in pancreatitis. Proc Natl Acad Sci U S A 102: 14386-14391, 2005. PMID: 16186498.
- Ito K, Miyashita Y, and Kasai H. Micromolar and submicromolar Ca2+ spikes regulating distinct cellular functions in pancreatic acinar cells. EMBO J 16: 242-251, 1997. PMID: 9029145.
- Iwatsuki N, and Petersen OH. Electrical coupling and uncoupling of exocrine acinar cells. J Cell Biol 79: 533-545, 1978. PMID: 569159.
- Jin K, Imada T, Nakamura S, Izuta Y, Oonishi E, Shibuya M, Sakaguchi H, Adachi T, and Tsubota K. Intravital Two-photon Imaging of Ca(2+) signaling in Secretory Organs of Yellow Cameleon Transgenic Mice. Sci Rep 8: 15880, 2018. PMID: 30367106.
- Kasai H, and Augustine GJ. Cytosolic Ca2+ gradients triggering unidirectional fluid secretion from exocrine pancreas. Nature 348: 735-738, 1990. PMID: 1701852.
- Kasai H, Li YX, and Miyashita Y. Subcellular distribution of Ca2+ release channels underlying Ca2+ waves and oscillations in exocrine pancreas. Cell 74: 669-677, 1993. PMID: 8395348.
- Kidd JF, Fogarty KE, Tuft RA, and Thorn P. The role of Ca2+ feedback in shaping InsP3-evoked Ca2+ signals in mouse pancreatic acinar cells. J Physiol 520 Pt 1: 187-201, 1999. PMID: 10517811.
- Kim MS, Hong JH, Li Q, Shin DM, Abramowitz J, Birnbaumer L, and Muallem S. Deletion of TRPC3 in mice reduces store-operated Ca2+ influx and the severity of acute pancreatitis. Gastroenterology 137: 1509-1517, 2009. PMID: 19622358.
- Kirichok Y, Krapivinsky G, and Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427: 360-364, 2004. PMID: 14737170.
- Krause E, Gobel A, and Schulz I. Cell side-specific sensitivities of intracellular Ca2+ stores for inositol 1,4,5-trisphosphate, cyclic ADP-ribose, and nicotinic acid adenine dinucleotide phosphate in permeabilized pancreatic acinar cells from mouse. J Biol Chem 277: 11696-11702, 2002. PMID: 11809747.
- Kruger B, Albrecht E, and Lerch MM. The role of intracellular calcium signaling in premature protease activation and the onset of pancreatitis. Am J Pathol 157: 43-50, 2000. PMID: 10880374.
- LeBeau AP, Yule DI, Groblewski GE, and Sneyd J. Agonist-dependent phosphorylation of the inositol 1,4,5-trisphosphate receptor: A possible mechanism for agonist-specific calcium oscillations in pancreatic acinar cells. J Gen Physiol 113: 851-872, 1999. PMID: 10352035.
- Lee HC, Aarhus R, and Graeff RM. Sensitization of calcium-induced calcium release by cyclic ADP-ribose and calmodulin. J Biol Chem 270: 9060-9066, 1995. PMID: 7721819.
- Lee MG, Xu X, Zeng W, Diaz J, Kuo TH, Wuytack F, Racymaekers L, and Muallem S. Polarized expression of Ca2+ pumps in pancreatic and salivary gland cells. Role in initiation and propagation of [Ca2+]i waves. J Biol Chem 272: 15771-15776, 1997. PMID: 9188473.
- Lee MG, Xu X, Zeng W, Diaz J, Wojcikiewicz RJ, Kuo TH, Wuytack F, Racymaekers L, and Muallem S. Polarized expression of Ca2+ channels in pancreatic and salivary gland cells. Correlation with initiation and propagation of [Ca2+]i waves. J Biol Chem 272: 15765-15770, 1997. PMID: 9188472.
- Leite MF, Burgstahler AD, and Nathanson MH. Ca2+ waves require sequential activation of inositol trisphosphate receptors and ryanodine receptors in pancreatic acini. Gastroenterology 122: 415-427, 2002. PMID: 11832456.
- Leite MF, Dranoff JA, Gao L, and Nathanson MH. Expression and subcellular localization of the ryanodine receptor in rat pancreatic acinar cells. Biochem J 337 ( Pt 2): 305-309, 1999. PMID: 9882629.
- Lewarchik CM, Orabi AI, Jin S, Wang D, Muili KA, Shah AU, Eisses JF, Malik A, Bottino R, Jayaraman T, and Husain SZ. The ryanodine receptor is expressed in human pancreatic acinar cells and contributes to acinar cell injury. Am J Physiol Gastrointest Liver Physiol 307: G574-581, 2014. PMID: 25012845.
- Li Q, Luo X, and Muallem S. Functional mapping of Ca2+ signaling complexes in plasma membrane microdomains of polarized cells. J Biol Chem 279: 27837-27840, 2004. PMID: 15123684.
- Lin-Moshier Y, Walseth TF, Churamani D, Davidson SM, Slama JT, Hooper R, Brailoiu E, Patel S, and Marchant JS. Photoaffinity labeling of nicotinic acid adenine dinucleotide phosphate (NAADP) targets in mammalian cells. J Biol Chem 287: 2296-2307, 2012. PMID: 22117075.
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr., and Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol 15: 1235-1241, 2005. PMID: 16005298.
- Liu X, Cheng KT, Bandyopadhyay BC, Pani B, Dietrich A, Paria BC, Swaim WD, Beech D, Yildrim E, Singh BB, Birnbaumer L, and Ambudkar IS. Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1(-/-) mice. Proc Natl Acad Sci U S A 104: 17542-17547, 2007. PMID: 17956991.
- Lugea A, Waldron RT, Mareninova OA, Shalbueva N, Deng N, Su HY, Thomas DD, Jones EK, Messenger SW, Yang J, Hu C, Gukovsky I, Liu Z, Groblewski GE, Gukovskaya AS, Gorelick FS, and Pandol SJ. Human Pancreatic Acinar Cells: Proteomic Characterization, Physiologic Responses, and Organellar Disorders in ex Vivo Pancreatitis. Am J Pathol 187: 2726-2743, 2017. PMID: 28935577.
- Luik RM, Wu MM, Buchanan J, and Lewis RS. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol 174: 815-825, 2006. PMID: 16966423.
- Luo X, Popov S, Bera AK, Wilkie TM, and Muallem S. RGS proteins provide biochemical control of agonist-evoked [Ca2+]i oscillations. Mol Cell 7: 651-660, 2001. PMID: 11463389.
- Lur G, Haynes LP, Prior IA, Gerasimenko OV, Feske S, Petersen OH, Burgoyne RD, and Tepikin AV. Ribosome-free terminals of rough ER allow formation of STIM1 puncta and segregation of STIM1 from IP(3) receptors. Curr Biol 19: 1648-1653, 2009. PMID: 19765991.
- Mak DO, and Foskett JK. Single-channel inositol 1,4,5-trisphosphate receptor currents revealed by patch clamp of isolated Xenopus oocyte nuclei. J Biol Chem 269: 29375-29378, 1994. PMID: 7961913.
- Maranto AR. Primary structure, ligand binding, and localization of the human type 3 inositol 1,4,5-trisphosphate receptor expressed in intestinal epithelium. J Biol Chem 269: 1222-1230, 1994. PMID: 8288584.
- Matozaki T, Sakamoto C, Nishisaki H, Suzuki T, Wada K, Matsuda K, Nakano O, Konda Y, Nagao M, and Kasuga M. Cholecystokinin inhibits phosphatidylcholine synthesis via a Ca(2+)-calmodulin-dependent pathway in isolated rat pancreatic acini. A possible mechanism for diacylglycerol accumulation. J Biol Chem 266: 22246-22253, 1991. PMID: 1657994.
- Matozaki T, Zhu WY, Tsunoda Y, Goke B, and Williams JA. Intracellular mediators of bombesin action on rat pancreatic acinar cells. Am J Physiol 260: G858-864, 1991. PMID: 1711779.
- Meda P, Findlay I, Kolod E, Orci L, and Petersen OH. Short and reversible uncoupling evokes little change in the gap junctions of pancreatic acinar cells. J Ultrastruct Res 83: 69-84, 1983. PMID: 6406682.
- Metz DC, Patto RJ, Mrozinski JE, Jr., Jensen RT, Turner RJ, and Gardner JD. Thapsigargin defines the roles of cellular calcium in secretagogue-stimulated enzyme secretion from pancreatic acini. J Biol Chem 267: 20620-20629, 1992. PMID: 1383205.
- Mignen O, Thompson JL, Yule DI, and Shuttleworth TJ. Agonist activation of arachidonate-regulated Ca2+-selective (ARC) channels in murine parotid and pancreatic acinar cells. J Physiol 564: 791-801, 2005. PMID: 15760932.
- Mignery GA, Newton CL, Archer BT, 3rd, and Sudhof TC. Structure and expression of the rat inositol 1,4,5-trisphosphate receptor. J Biol Chem 265: 12679-12685, 1990. PMID: 2165071.
- Mogami H, Nakano K, Tepikin AV, and Petersen OH. Ca2+ flow via tunnels in polarized cells: recharging of apical Ca2+ stores by focal Ca2+ entry through basal membrane patch. Cell 88: 49-55, 1997. PMID: 9019404.
- Mogami H, Tepikin AV, and Petersen OH. Termination of cytosolic Ca2+ signals: Ca2+ reuptake into intracellular stores is regulated by the free Ca2+ concentration in the store lumen. EMBO J 17: 435-442, 1998. PMID: 9430635.
- Muili KA, Ahmad M, Orabi AI, Mahmood SM, Shah AU, Molkentin JD, and Husain SZ. Pharmacological and genetic inhibition of calcineurin protects against carbachol-induced pathological zymogen activation and acinar cell injury. Am J Physiol Gastrointest Liver Physiol 302: G898-905, 2012. PMID: 22323127.
- Muili KA, Wang D, Orabi AI, Sarwar S, Luo Y, Javed TA, Eisses JF, Mahmood SM, Jin S, Singh VP, Ananthanaravanan M, Perides G, Williams JA, Molkentin JD, and Husain SZ. Bile acids induce pancreatic acinar cell injury and pancreatitis by activating calcineurin. J Biol Chem 288: 570-580, 2013. PMID: 23148215.
- Nagata J, Guerra MT, Shugrue CA, Gomes DA, Nagata N, and Nathanson MH. Lipid rafts establish calcium waves in hepatocytes. Gastroenterology 133: 256-267, 2007. PMID: 17631147.
- Nathanson MH, Fallon MB, Padfield PJ, and Maranto AR. Localization of the type 3 inositol 1,4,5-trisphosphate receptor in the Ca2+ wave trigger zone of pancreatic acinar cells. J Biol Chem 269: 4693-4696, 1994. PMID: 7508924.
- Nathanson MH, Padfield PJ, O'Sullivan AJ, Burgstahler AD, and Jamieson JD. Mechanism of Ca2+ wave propagation in pancreatic acinar cells. J Biol Chem 267: 18118-18121, 1992. PMID: 1517244.
- Ong HL, Cheng KT, Liu X, Bandyopadhyay BC, Paria BC, Soboloff J, Pani B, Gwack Y, Srikanth S, Singh BB, Gill DL, and Ambudkar IS. Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J Biol Chem 282: 9105-9116, 2007. PMID: 17224452.
- Orabi AI, Luo Y, Ahmad MU, Shah AU, Mannan Z, Wang D, Sarwar S, Muili KA, Shugrue C, Kolodecik TR, Singh VP, Lowe ME, Thrower E, Chen J, and Husain SZ. IP3 receptor type 2 deficiency is associated with a secretory defect in the pancreatic acinar cell and an accumulation of zymogen granules. PLoS One 7: e48465, 2012. PMID: 23185258.
- Oshima Y, Imamura T, Shintani A, Kajiura-Kobayashi H, Hibi T, Nagai T, Nonaka S, and Nemoto T. Ultrasensitive imaging of Ca2+ dynamics in pancreatic acinar cells of yellow cameleon-nano transgenic mice. Int J Mol Sci 15: 19971-19986, 2014. PMID: 25372943.
- Osipchuk YV, Wakui M, Yule DI, Gallacher DV, and Petersen OH. Cytoplasmic Ca2+ oscillations evoked by receptor stimulation, G-protein activation, internal application of inositol trisphosphate or Ca2+: simultaneous microfluorimetry and Ca2+ dependent Cl- current recording in single pancreatic acinar cells. EMBO J 9: 697-704, 1990. PMID: 1690123.
- Ozawa T. Elucidation of the ryanodine-sensitive Ca2+ release mechanism of rat pancreatic acinar cells: modulation by cyclic ADP-ribose and FK506. Biochim Biophys Acta 1693: 159-166, 2004. PMID: 15363629.
- Pandol SJ, Schoeffield MS, Fimmel CJ, and Muallem S. The agonist-sensitive calcium pool in the pancreatic acinar cell. Activation of plasma membrane Ca2+ influx mechanism. J Biol Chem 262: 16963-16968, 1987. PMID: 3680283.
- Park HS, Betzenhauser MJ, Won JH, Chen J, and Yule DI. The type 2 inositol (1,4,5)-trisphosphate (InsP3) receptor determines the sensitivity of InsP3-induced Ca2+ release to ATP in pancreatic acinar cells. J Biol Chem 283: 26081-26088, 2008. PMID: 18658132.
- Park MK, Ashby MC, Erdemli G, Petersen OH, and Tepikin AV. Perinuclear, perigranular and sub-plasmalemmal mitochondria have distinct functions in the regulation of cellular calcium transport. EMBO J 20: 1863-1874, 2001. PMID: 11296220.
- Park MK, Lomax RB, Tepikin AV, and Petersen OH. Local uncaging of caged Ca(2+) reveals distribution of Ca(2+)-activated Cl(-) channels in pancreatic acinar cells. Proc Natl Acad Sci U S A 98: 10948-10953, 2001. PMID: 11535807.
- Park MK, Petersen OH, and Tepikin AV. The endoplasmic reticulum as one continuous Ca(2+) pool: visualization of rapid Ca(2+) movements and equilibration. EMBO J 19: 5729-5739, 2000. PMID: 11060024.
- Patel S, Joseph SK, and Thomas AP. Molecular properties of inositol 1,4,5-trisphosphate receptors. Cell Calcium 25: 247-264, 1999. PMID: 10378086.
- Patterson RL, Boehning D, and Snyder SH. Inositol 1,4,5-trisphosphate receptors as signal integrators. Annu Rev Biochem 73: 437-465, 2004. PMID: 15189149.
- Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, and Mootha VK. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 467: 291-296, 2010. PMID: 20693986.
- Petersen CC, Toescu EC, and Petersen OH. Different patterns of receptor-activated cytoplasmic Ca2+ oscillations in single pancreatic acinar cells: dependence on receptor type, agonist concentration and intracellular Ca2+ buffering. EMBO J 10: 527-533, 1991. PMID: 1705883.
- Pieper AA, Brat DJ, O'Hearn E, Krug DK, Kaplin AI, Takahashi K, Greenberg JH, Ginty D, Molliver ME, and Snyder SH. Differential neuronal localizations and dynamics of phosphorylated and unphosphorylated type 1 inositol 1,4,5-trisphosphate receptors. Neuroscience 102: 433-444, 2001. PMID: 11166129.
- Ponnappa BC, Dormer RL, and Williams JA. Characterization of an ATP-dependent Ca2+ uptake system in mouse pancreatic microsomes. Am J Physiol 240: G122-129, 1981. PMID: 6258447.
- Pralong WF, Wollheim CB, and Bruzzone R. Measurement of cytosolic free Ca2+ in individual pancreatic acini. FEBS Lett 242: 79-84, 1988. PMID: 2462514.
- Putney JW, Jr. A model for receptor-regulated calcium entry. Cell Calcium 7: 1-12, 1986. PMID: 2420465.
- Raraty M, Ward J, Erdemli G, Vaillant C, Neoptolemos JP, Sutton R, and Petersen OH. Calcium-dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc Natl Acad Sci U S A 97: 13126-13131, 2000. PMID: 11087863.
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, and Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280: 1763-1766, 1998. PMID: 9624056.
- Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, and Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol 169: 435-445, 2005. PMID: 15866891.
- Rosenzweig SA, Miller LJ, and Jamieson JD. Identification and localization of cholecystokinin-binding sites on rat pancreatic plasma membranes and acinar cells: a biochemical and autoradiographic study. J Cell Biol 96: 1288-1297, 1983. PMID: 6841449.
- Ruas M, Davis LC, Chen CC, Morgan AJ, Chuang KT, Walseth TF, Grimm C, Garnham C, Powell T, Platt N, Platt FM, Biel M, Wahl-Schott C, Parrington J, and Galione A. Expression of Ca2+-permeable two-pore channels rescues NAADP signalling in TPC-deficient cells. EMBO J 2015. PMID: 25872774.
- Seo MD, Velamakanni S, Ishiyama N, Stathopulos PB, Rossi AM, Khan SA, Dale P, Li C, Ames JB, Ikura M, and Taylor CW. Structural and functional conservation of key domains in InsP3 and ryanodine receptors. Nature 483: 108-112, 2012. PMID: 22286060.
- Shin DM, Luo X, Wilkie TM, Miller LJ, Peck AB, Humphreys-Beher MG, and Muallem S. Polarized expression of G protein-coupled receptors and an all-or-none discharge of Ca2+ pools at initiation sites of [Ca2+]i waves in polarized exocrine cells. J Biol Chem 276: 44146-44156, 2001. PMID: 11553617.
- Shuttleworth TJ. Orai channels - new insights, new ideas. J Physiol 590: 4155-4156, 2012. PMID: 22962035.
- Sitsapesan R, McGarry SJ, and Williams AJ. Cyclic ADP-ribose, the ryanodine receptor and Ca2+ release. Trends Pharmacol Sci 16: 386-391, 1995. PMID: 8578608.
- Sjodin L, Dahlen HG, and Gylfe E. Calcium oscillations in guinea-pig pancreatic acinar cells exposed to carbachol, cholecystokinin and substance P. J Physiol 444: 763-776, 1991. PMID: 1726599.
- Sneyd J, Tsaneva-Atanasova K, Reznikov V, Bai Y, Sanderson MJ, and Yule DI. A method for determining the dependence of calcium oscillations on inositol trisphosphate oscillations. Proc Natl Acad Sci U S A 103: 1675-1680, 2006. PMID: 16446452.
- Stauffer PL, Zhao H, Luby-Phelps K, Moss RL, Star RA, and Muallem S. Gap junction communication modulates [Ca2+]i oscillations and enzyme secretion in pancreatic acini. J Biol Chem 268: 19769-19775, 1993. PMID: 8366115.
- Sternfeld L, Krause E, Guse AH, and Schulz I. Hormonal control of ADP-ribosyl cyclase activity in pancreatic acinar cells from rats. J Biol Chem 278: 33629-33636, 2003. PMID: 12807891.
- Straub SV, Giovannucci DR, Bruce JI, and Yule DI. A role for phosphorylation of inositol 1,4,5-trisphosphate receptors in defining calcium signals induced by Peptide agonists in pancreatic acinar cells. J Biol Chem 277: 31949-31956, 2002. PMID: 12065595.
- Straub SV, Giovannucci DR, and Yule DI. Calcium wave propagation in pancreatic acinar cells: functional interaction of inositol 1,4,5-trisphosphate receptors, ryanodine receptors, and mitochondria. J Gen Physiol 116: 547-560, 2000. PMID: 11004204.
- Streb H, Bayerdorffer E, Haase W, Irvine RF, and Schulz I. Effect of inositol-1,4,5-trisphosphate on isolated subcellular fractions of rat pancreas. J Membr Biol 81: 241-253, 1984. PMID: 6334162.
- Streb H, Heslop JP, Irvine RF, Schulz I, and Berridge MJ. Relationship between secretagogue-induced Ca2+ release and inositol polyphosphate production in permeabilized pancreatic acinar cells. J Biol Chem 260: 7309-7315, 1985. PMID: 3997871.
- Streb H, Irvine RF, Berridge MJ, and Schulz I. Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature 306: 67-69, 1983. PMID: 6605482.
- Stuenkel EL, Tsunoda Y, and Williams JA. Secretagogue induced calcium mobilization in single pancreatic acinar cells. Biochem Biophys Res Commun 158: 863-869, 1989. PMID: 2920043.
- Takasawa S, Nata K, Yonekura H, and Okamoto H. Cyclic ADP-ribose in insulin secretion from pancreatic beta cells. Science 259: 370-373, 1993. PMID: 8420005.
- Taylor CW, Genazzani AA, and Morris SA. Expression of inositol trisphosphate receptors. Cell Calcium 26: 237-251, 1999. PMID: 10668562.
- Taylor CW, and Laude AJ. IP3 receptors and their regulation by calmodulin and cytosolic Ca2+. Cell Calcium 32: 321-334, 2002. PMID: 12543092.
- Tepikin AV, Llopis J, Snitsarev VA, Gallacher DV, and Petersen OH. The droplet technique: measurement of calcium extrusion from single isolated mammalian cells. Pflugers Arch 428: 664-670, 1994. PMID: 7838690.
- Tepikin AV, Voronina SG, Gallacher DV, and Petersen OH. Acetylcholine-evoked increase in the cytoplasmic Ca2+ concentration and Ca2+ extrusion measured simultaneously in single mouse pancreatic acinar cells. J Biol Chem 267: 3569-3572, 1992. PMID: 1310974.
- Tepikin AV, Voronina SG, Gallacher DV, and Petersen OH. Pulsatile Ca2+ extrusion from single pancreatic acinar cells during receptor-activated cytosolic Ca2+ spiking. J Biol Chem 267: 14073-14076, 1992. PMID: 1629206.
- Thompson JL, and Shuttleworth TJ. Exploring the unique features of the ARC channel, a store-independent Orai channel. Channels (Austin) 7: 364-373, 2013. PMID: 24025406.
- Thompson JL, and Shuttleworth TJ. Molecular basis of activation of the arachidonate-regulated Ca2+ (ARC) channel, a store-independent Orai channel, by plasma membrane STIM1. J Physiol 591: 3507-3523, 2013. PMID: 23690558.
- Thorn P, Gerasimenko O, and Petersen OH. Cyclic ADP-ribose regulation of ryanodine receptors involved in agonist evoked cytosolic Ca2+ oscillations in pancreatic acinar cells. EMBO J 13: 2038-2043, 1994. PMID: 7514529.
- Thorn P, Lawrie AM, Smith PM, Gallacher DV, and Petersen OH. Local and global cytosolic Ca2+ oscillations in exocrine cells evoked by agonists and inositol trisphosphate. Cell 74: 661-668, 1993. PMID: 8395347.
- Tinel H, Cancela JM, Mogami H, Gerasimenko JV, Gerasimenko OV, Tepikin AV, and Petersen OH. Active mitochondria surrounding the pancreatic acinar granule region prevent spreading of inositol trisphosphate-evoked local cytosolic Ca(2+) signals. EMBO J 18: 4999-5008, 1999. PMID: 10487752.
- Toescu EC, Lawrie AM, Petersen OH, and Gallacher DV. Spatial and temporal distribution of agonist-evoked cytoplasmic Ca2+ signals in exocrine acinar cells analysed by digital image microscopy. EMBO J 11: 1623-1629, 1992. PMID: 1563357.
- Toescu EC, O'Neill SC, Petersen OH, and Eisner DA. Caffeine inhibits the agonist-evoked cytosolic Ca2+ signal in mouse pancreatic acinar cells by blocking inositol trisphosphate production. J Biol Chem 267: 23467-23470, 1992. PMID: 1429689.
- Tsunoda Y, Stuenkel EL, and Williams JA. Characterization of sustained [Ca2+]i increase in pancreatic acinar cells and its relation to amylase secretion. Am J Physiol 259: G792-801, 1990. PMID: 1700626.
- Tsunoda Y, Stuenkel EL, and Williams JA. Oscillatory mode of calcium signaling in rat pancreatic acinar cells. Am J Physiol 258: C147-155, 1990. PMID: 2301562.
- Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, and Kinet JP. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312: 1220-1223, 2006. PMID: 16645049.
- Voronina S, Collier D, Chvanov M, Middlehurst B, Beckett AJ, Prior IA, Criddle DN, Begg M, Mikoshiba K, Sutton R, and Tepikin AV. The role of Ca2+ influx in endocytic vacuole formation in pancreatic acinar cells. Biochem J 465: 405-412, 2015. PMID: 25370603.
- Voronina S, Sukhomlin T, Johnson PR, Erdemli G, Petersen OH, and Tepikin A. Correlation of NADH and Ca2+ signals in mouse pancreatic acinar cells. J Physiol 539: 41-52, 2002. PMID: 11850500.
- Voronina S, and Tepikin A. Mitochondrial calcium in the life and death of exocrine secretory cells. Cell Calcium 52: 86-92, 2012. PMID: 22571865.
- Voronina SG, Barrow SL, Simpson AW, Gerasimenko OV, da Silva Xavier G, Rutter GA, Petersen OH, and Tepikin AV. Dynamic changes in cytosolic and mitochondrial ATP levels in pancreatic acinar cells. Gastroenterology 138: 1976-1987, 2010. PMID: 20102715.
- Wagner LE, 2nd, and Yule DI. Differential regulation of the InsP(3) receptor type-1 and -2 single channel properties by InsP(3), Ca(2)(+) and ATP. J Physiol 590: 3245-3259, 2012. PMID: 22547632.
- Wakasugi H, Kimura T, Haase W, Kribben A, Kaufmann R, and Schulz I. Calcium uptake into acini from rat pancreas: evidence for intracellular ATP-dependent calcium sequestration. J Membr Biol 65: 205-220, 1982. PMID: 6801263.
- Wakui M, Potter BV, and Petersen OH. Pulsatile intracellular calcium release does not depend on fluctuations in inositol trisphosphate concentration. Nature 339: 317-320, 1989. PMID: 2498663.
- Waldron RT, Chen Y, Pham H, Go A, Su HY, Hu C, Wen L, Husain SZ, Sugar CA, Roos J, Ramos S, Lugea A, Dunn M, Stauderman K, and Pandol SJ. The Orai Ca(2+) channel inhibitor CM4620 targets both parenchymal and immune cells to reduce inflammation in experimental acute pancreatitis. J Physiol 597: 3085-3105, 2019. PMID: 31050811.
- Walseth TF, Lin-Moshier Y, Jain P, Ruas M, Parrington J, Galione A, Marchant JS, and Slama JT. Photoaffinity labeling of high affinity nicotinic acid adenine dinucleotide phosphate (NAADP)-binding proteins in sea urchin egg. J Biol Chem 287: 2308-2315, 2012. PMID: 22117077.
- Wen L, Voronina S, Javed MA, Awais M, Szatmary P, Latawiec D, Chvanov M, Collier D, Huang W, Barrett J, Begg M, Stauderman K, Roos J, Grigoryev S, Ramos S, Rogers E, Whitten J, Velicelebi G, Dunn M, Tepikin AV, Criddle DN, and Sutton R. Inhibitors of ORAI1 Prevent Cytosolic Calcium-Associated Injury of Human Pancreatic Acinar Cells and Acute Pancreatitis in 3 Mouse Models. Gastroenterology 149: 481-492 e487, 2015. PMID: 25917787.
- Willems PH, van Nooij IG, Haenen HE, and de Pont JJ. Phorbol ester inhibits cholecystokinin octapeptide-induced amylase secretion and calcium mobilization, but is without effect on secretagogue-induced hydrolysis of phosphatidylinositol 4,5-bisphosphate in rabbit pancreatic acini. Biochim Biophys Acta 930: 230-236, 1987. PMID: 2441762.
- Wojcikiewicz RJ. Type I, II, and III inositol 1,4,5-trisphosphate receptors are unequally susceptible to down-regulation and are expressed in markedly different proportions in different cell types. J Biol Chem 270: 11678-11683, 1995. PMID: 7744807.
- Wojcikiewicz RJ, Ernst SA, and Yule DI. Secretagogues cause ubiquitination and down-regulation of inositol 1, 4,5-trisphosphate receptors in rat pancreatic acinar cells. Gastroenterology 116: 1194-1201, 1999. PMID: 10220512.
- Worley PF, Zeng W, Huang GN, Yuan JP, Kim JY, Lee MG, and Muallem S. TRPC channels as STIM1-regulated store-operated channels. Cell Calcium 42: 205-211, 2007. PMID: 17517433.
- Wu MM, Buchanan J, Luik RM, and Lewis RS. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol 174: 803-813, 2006. PMID: 16966422.
- Xu X, Zeng W, and Muallem S. Regulation of the inositol 1,4,5-trisphosphate-activated Ca2+ channel by activation of G proteins. J Biol Chem 271: 11737-11744, 1996. PMID: 8662624.
- Xu X, Zeng W, Popov S, Berman DM, Davignon I, Yu K, Yowe D, Offermanns S, Muallem S, and Wilkie TM. RGS proteins determine signaling specificity of Gq-coupled receptors. J Biol Chem 274: 3549-3556, 1999. PMID: 9920901.
- Yamasaki M, Masgrau R, Morgan AJ, Churchill GC, Patel S, Ashcroft SJ, and Galione A. Organelle selection determines agonist-specific Ca2+ signals in pancreatic acinar and beta cells. J Biol Chem 279: 7234-7240, 2004. PMID: 14660554.
- Yamasaki M, Thomas JM, Churchill GC, Garnham C, Lewis AM, Cancela JM, Patel S, and Galione A. Role of NAADP and cADPR in the induction and maintenance of agonist-evoked Ca2+ spiking in mouse pancreatic acinar cells. Curr Biol 15: 874-878, 2005. PMID: 15886108.
- Yano K, Petersen OH, and Tepikin AV. Dual sensitivity of sarcoplasmic/endoplasmic Ca2+-ATPase to cytosolic and endoplasmic reticulum Ca2+ as a mechanism of modulating cytosolic Ca2+ oscillations. Biochem J 383: 353-360, 2004. PMID: 15260801.
- Yule DI, Betzenhauser MJ, and Joseph SK. Linking structure to function: Recent lessons from inositol 1,4,5-trisphosphate receptor mutagenesis. Cell Calcium 47: 469-479, 2010. PMID: 20510450.
- Yule DI, Ernst SA, Ohnishi H, and Wojcikiewicz RJ. Evidence that zymogen granules are not a physiologically relevant calcium pool. Defining the distribution of inositol 1,4,5-trisphosphate receptors in pancreatic acinar cells. J Biol Chem 272: 9093-9098, 1997. PMID: 9083036.
- Yule DI, and Gallacher DV. Oscillations of cytosolic calcium in single pancreatic acinar cells stimulated by acetylcholine. FEBS Lett 239: 358-362, 1988. PMID: 3141216.
- Yule DI, Kim ET, and Williams JA. Tyrosine kinase inhibitors attenuate "capacitative" Ca2+ influx in rat pancreatic acinar cells. Biochem Biophys Res Commun 202: 1697-1704, 1994. PMID: 8060359.
- Yule DI, Lawrie AM, and Gallacher DV. Acetylcholine and cholecystokinin induce different patterns of oscillating calcium signals in pancreatic acinar cells. Cell Calcium 12: 145-151, 1991. PMID: 2059990.
- Yule DI, Stuenkel E, and Williams JA. Intercellular calcium waves in rat pancreatic acini: mechanism of transmission. Am J Physiol 271: C1285-1294, 1996. PMID: 8897836.
- Yule DI, and Williams JA. Mastoparan induces oscillations of cytosolic Ca2+ in rat pancreatic acinar cells. Biochem Biophys Res Commun 177: 159-165, 1991. PMID: 2043103.
- Zariwala HA, Borghuis BG, Hoogland TM, Madisen L, Tian L, De Zeeuw CI, Zeng H, Looger LL, Svoboda K, and Chen TW. A Cre-dependent GCaMP3 reporter mouse for neuronal imaging in vivo. J Neurosci 32: 3131-3141, 2012. PMID: 22378886.
- Zeng W, Mak DO, Li Q, Shin DM, Foskett JK, and Muallem S. A new mode of Ca2+ signaling by G protein-coupled receptors: gating of IP3 receptor Ca2+ release channels by Gbetagamma. Curr Biol 13: 872-876, 2003. PMID: 12747838.
- Zeng W, Xu X, and Muallem S. Gbetagamma transduces [Ca2+]i oscillations and Galphaq a sustained response during stimulation of pancreatic acinar cells with [Ca2+]i-mobilizing agonists. J Biol Chem 271: 18520-18526, 1996. PMID: 8702499.
- Zeng W, Xu X, Popov S, Mukhopadhyay S, Chidiac P, Swistok J, Danho W, Yagaloff KA, Fisher SL, Ross EM, Muallem S, and Wilkie TM. The N-terminal domain of RGS4 confers receptor-selective inhibition of G protein signaling. J Biol Chem 273: 34687-34690, 1998. PMID: 9856989.
- Zhang BX, Zhao H, Loessberg P, and Muallem S. Activation of the plasma membrane Ca2+ pump during agonist stimulation of pancreatic acini. J Biol Chem 267: 15419-15425, 1992. PMID: 1322397.
- Zhang BX, Zhao H, Loessberg PA, and Muallem S. Regulation of agonist-evoked [Ca2+]i oscillation by intracellular Ca2+ and Ba2+ in AR42J cells. Am J Physiol 262: C1125-1133, 1992. PMID: 1317097.
- Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, and Cahalan MD. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc Natl Acad Sci U S A 103: 9357-9362, 2006. PMID: 16751269.